Vogt-Koyanagi-Harada (VKH) sygdom
forfattere: Anthony P. Mai, BS; Charlene Tran, BS; Caroline P. Vilson, MD; Austin R. ræv, MD; H. Culver Boldt, MD
1.April 2019
indledende præsentation
hovedklager
sløret syn og hovedpine
nutidens historie sygdom
en 44-årig vietnamesisk kvinde præsenterede for akutafdelingen med en 10-dages historie med progressiv sløret syn i begge øjne og en tre-dages historie med svær hovedpine. Hendes centrale synstab var ikke forbedret med en brydning af hendes optiker. Hendes alvorlige occipital hovedpine forværredes med bevægelse og var forbundet med generaliseret utilpashed, ekstrem træthed, mild fotofobi og rive. Acetaminophen lindrede delvist smerten.
hun havde for nylig rejst til Vietnam, men nægtet at møde syge kontakter der. Hun benægtede kæbe claudication, feber, eller vægtændringer. Hun benægtede hududslæt, høreændringer, tinnitus, svimmelhed, følelsesløshed eller prikken. Hun nægtede nogensinde at have tuberkulose. Hun havde ingen tidligere synsproblemer, autoimmune tilstande eller kræft.
tidligere okulær historie
- historie med kosmetisk Øjenlågskirurgi (bilateral blepharoplasty) tre år tidligere
- ingen historie med okulært traume eller sygdom
tidligere medicinsk historie
medicin
Acetaminophen efter behov
allergier
ingen kendte lægemiddelallergier
familiehistorie
ingen historie med øjensygdom eller autoimmun sygdom
social historie
hun immigrerede fra Vietnam flere år før præsentationen. Hun er gift og har tre børn. Hun arbejder på en neglesalon. Hun spiser ikke tobaksvarer, alkohol eller ulovlige stoffer. Hun rejser til Vietnam hver sjette til tolv måned.
gennemgang af systemer
negativ bortset fra hvad der er detaljeret i historien om den nuværende sygdom
okulær undersøgelse
synsstyrke med / uden korrektion (Snellen)
- højre øje (OD): 20/300 (ingen forbedring med pinhole)
- venstre øje (OS): 20/60-2+2 (ingen forbedring med pinhole)
okulær motilitet / justering
fuld ekstraokulære bevægelser i begge øjne (OU)
intraokulært tryk (IOP): (Tonopen)
- OD: 12 mmHg
- OS: 14 mmHg
elever
- OD: 4 mm i mørke, 3 mm i lys, ingen relativ afferent pupilledefekt (RAPD)
- OS: 4 mm i mørke, 3 mm i lys, ingen RAPD
konfrontation visuel felter: (tæl fingre)
- OD: central scotoma
- os: total inferotemporal defekt
ekstern
normal på begge sider
spaltelampe eksamen
- låg/vipper: normal ou
- conjunctiva/sclera: klar og stille ou
- hornhinde: 1+ punkterede epiteliale erosioner, ingen keratiske udfældninger OU
- forreste kammer: Sporcelle og flare og dyb ou
- Iris: Normal arkitektur OU
- linse: klar OU
udvidet fundusundersøgelse (DFE)
- glasagtige: spor forreste glasagtige celler OU
- disk:
- OD: grad 3 skiveødem, hyperemisk
- os: grad 2-3 skiveødem, hyperemisk
- cup-to-Disc-forhold: 0,0 ou
- makula:
- od: 3 + cystoid makulært ødem (CME) og subretinalvæske (SRF), der strækker sig fra disken til den tidsmæssige makula. Ingen lipid eller ekssudater. Boggy-vises choroid.
- OS: 2 + CME og SRF strækker sig fra disken gennem fovea. 1-2 + lineær lipid strækker sig fra skive mod fovea. Boggy-vises choroid.
- fartøjer:
- OD: Beklædning midlertidigt
- OS: Normal
- periferi:
- od: cystisk retinal tuft anterior til ækvator ved 10:30
- OS: lav SRF anterior til ækvator ved 4:00
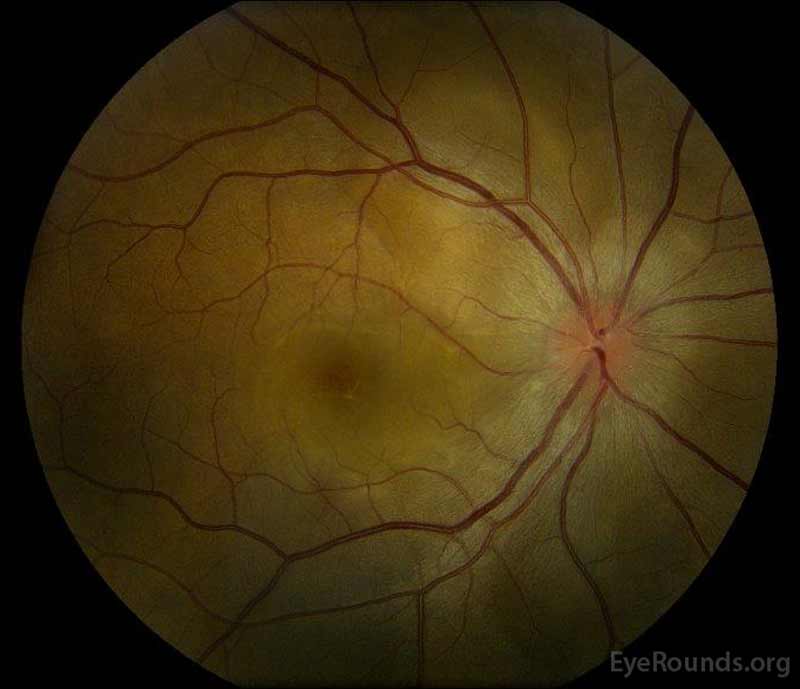 |
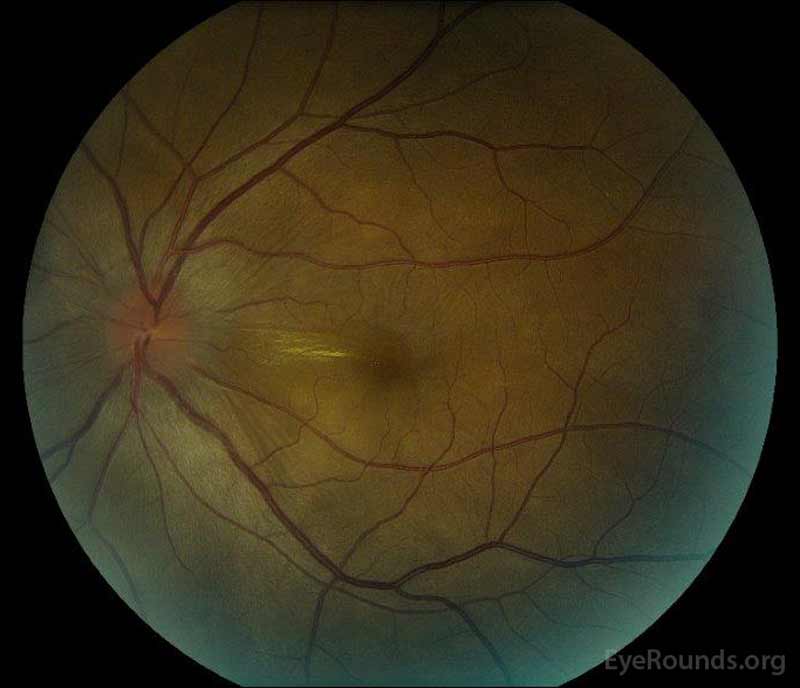 |
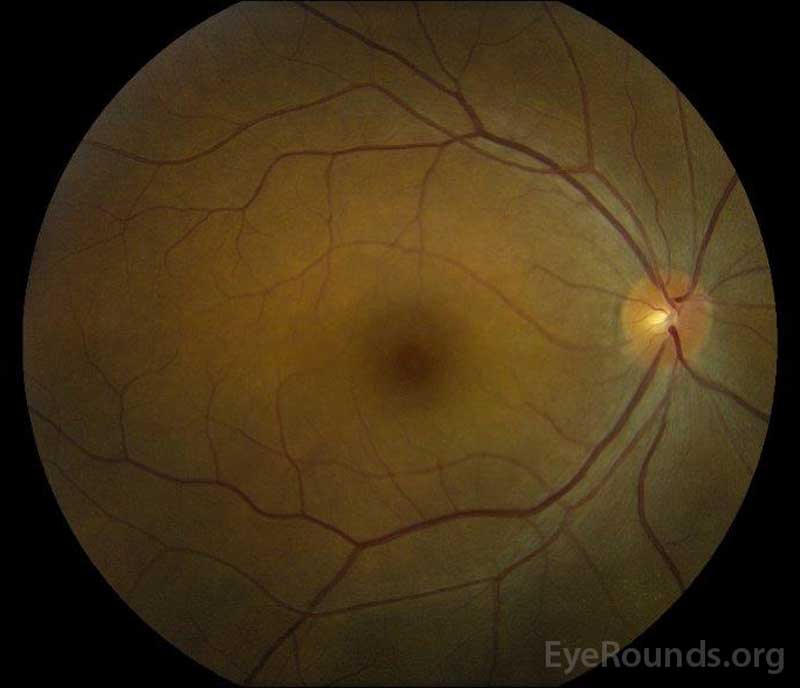
figur 1: farvefundusfotografier ved præsentation: (venstre billede) højre øje har skiveødem og mild hyperæmi samt subretinalvæske, der strækker sig fra disken midlertidigt gennem makulaen. Der er også en fokal serøs nethindeløsning superotemporal til disken langs den overlegne arkade. (Right image) The left eye has disc edema and mild hyperemia, along with subretinal fluid extending from the disc to the macula and linear lipid deposits in the nasal macula.
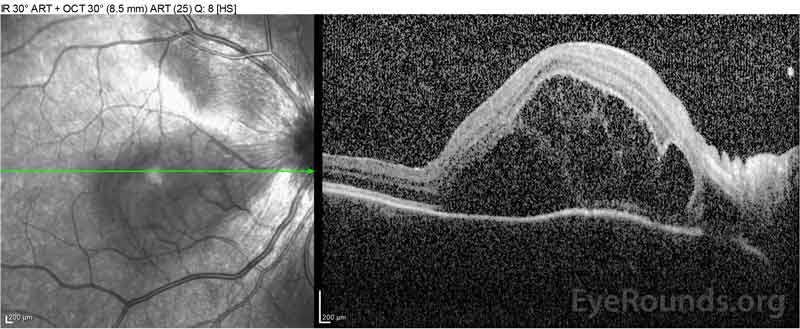 |
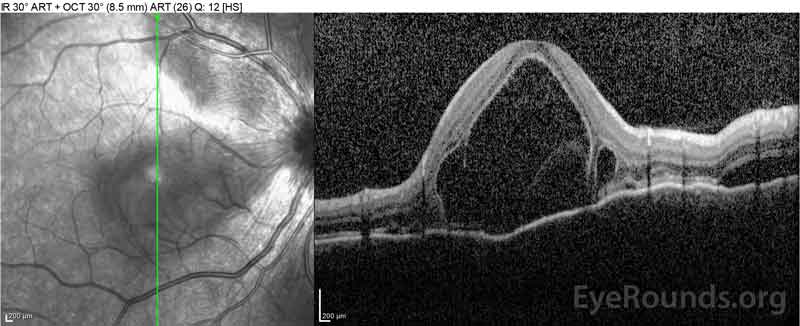 |
 |
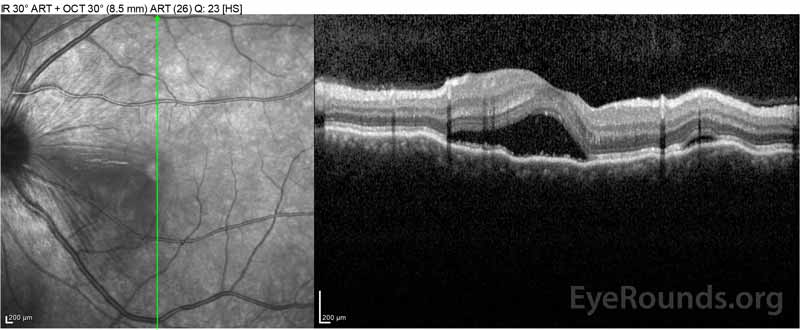 |
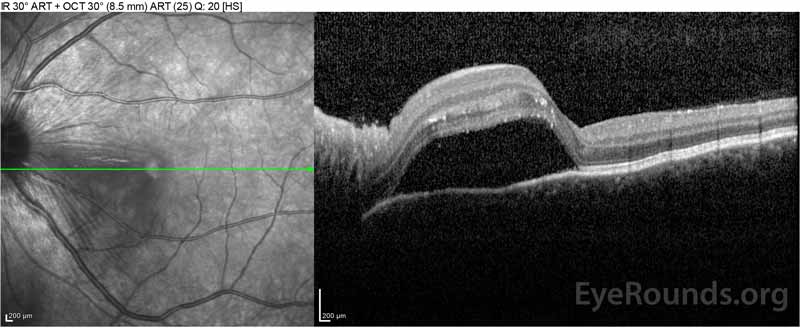
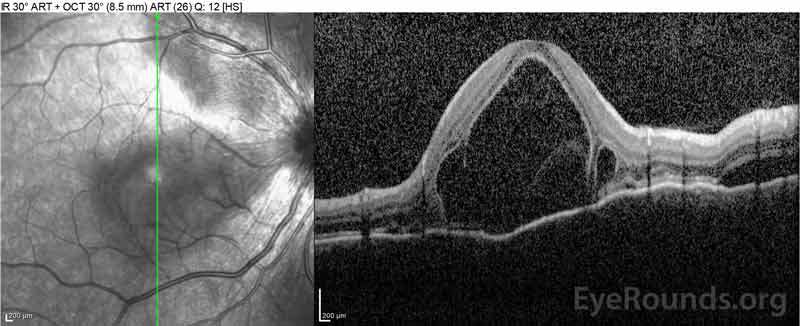
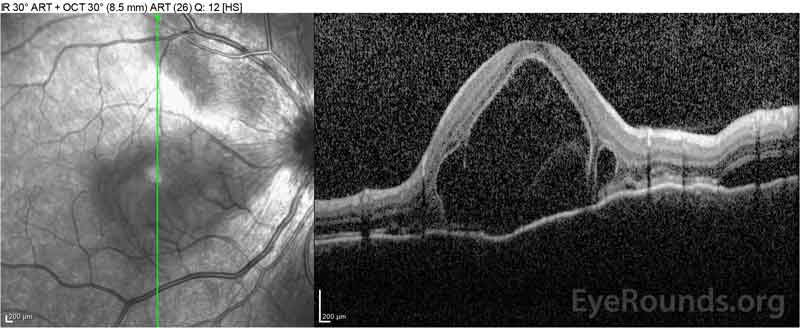
Figure 2: Optisk kohærens tomografi (OCT) i højre øje (øvre paneler) viser en serøs nethindeløsning, der involverer fovea med omfattende overliggende intraretinalvæske, forstyrrelse af de ydre nethindelag og bølger af den fortykkede choroid. OCT i venstre øje (bundpaneler) viser en serøs nethindeløsning i næsemakulaen, der strækker sig op til fovea.
 |
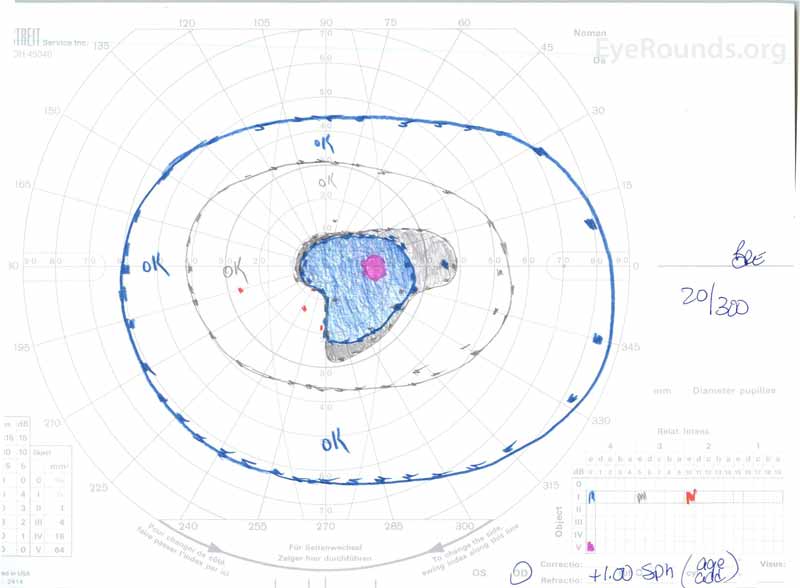 |
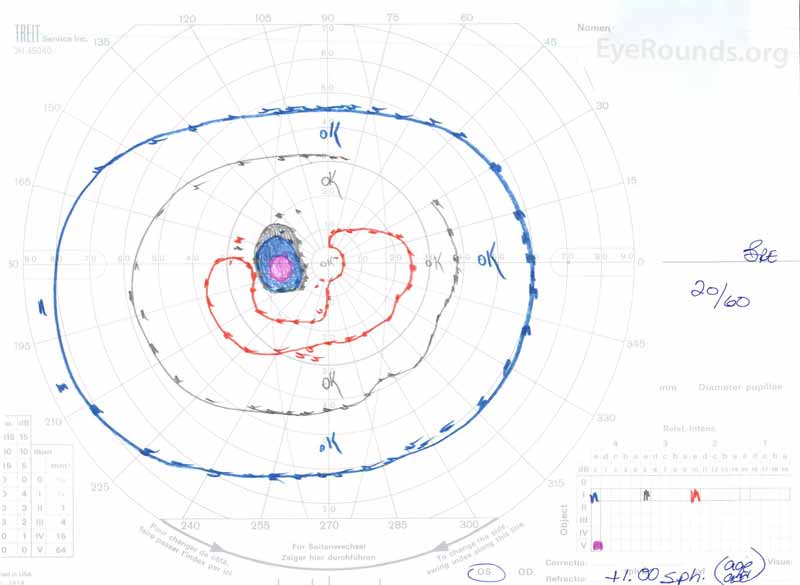
figur 3: Goldman visuelle felter (GVF), OU. (Venstre billede) OS viser en forstørret fysiologisk blind plet og mild central scotoma. (Højre billede) OD viser et moderat tæt centralt scotoma, der inkorporerer den fysiologiske blinde plet og strækker sig inferotemporalt.
B-scan: ingen tegn på skleritis, mild vitreal opacitet/celler inferiorly
differentialdiagnose
- akut posterior multifokal placoidpigmentepiteliopati (APMPPE)
- Central serøs chorioretinopati
- optisk neuritis
- Panuveitis
- autoimmun sygdom (f.eks. SLE, sarkoidose)
- infektion (e.g., syfilis, tuberkulose, Bartonella henselae)
- malignitet (f. eks. okulært lymfom)
- Posterior scleritis
- sympatisk ophthalmia
- Uveal effusion syndrom
- Vogt-Koyanagi-Harada syndrom
oparbejdning
komplet blodtælling
antal hvide blodlegemer: 4,9 k/mm3 (ref: 3,7-10,5)
antal røde blodlegemer 3,99 m/mm3 (ref: 4,0-5,2)
hæmoglobin 11,6 g/dl (Ref: 11,9-15,5)
hæmatokrit 35 % (ref:: 35-47)
Basic metabolic panel
Sodium 138 mEq/L (Ref: 135-145)
Potassium 4.3 mEq/L (Ref: 3.5-5.0)
Chloride 107 mEq/L (Ref: 95-107)
CO2 20 mEq/L (Ref: 22-29)
Blood urea nitrogen 16 mEq/dL (Ref: 10-20)
Creatinine 0.7 mg/dL (Ref: 0.5-1.0)
C-reactive Protein (CRP): <0.5 mg/dL (Ref: <=0.5)
Erythrocyte sedimentation rate (ESR): 12 mm/Hr (Ref: 0-20)
Angiotensin–converting enzyme (ACE): 13 U/L (Ref: 8-52)
kvantiferon-TB Gold: negativ
jern, blod 54 mikrogram/dL (Ref: 37-145)
Total jernbindingskapacitet 379 mikrogram/dL (Ref: 250-425)
klinisk forløb
patienten blev oprindeligt evalueret af akutafdelingen i betragtning af hendes klager over ny begyndelse alvorlig hovedpine og synstab. Hjernecomputertomografi (CT) og magnetisk resonansbilleddannelse (MRI) scanninger var ikke bemærkelsesværdige. ESR og CRP var inden for normale niveauer. Ophthalmology clinic evaluerede hende den følgende dag og fandt bilaterale serøse nethindeafdelinger og panuveitis. ACE og kvantiferon-TB Gold labs var begge negative. Hun blev diagnosticeret med Vogt-Koyanagi-Harada sygdom baseret på hendes kliniske præsentation og asiatiske afstamning. Hun blev behandlet med 80 mg prednison dagligt, acetaminophen efter behov for hovedpine og vitamin D og calciumtilskud. Hendes hovedpine hurtigt løst, og hendes synsstyrke støt forbedret i løbet af de følgende to uger. Hendes prednison dosering blev derefter tilspidset ned til 40 mg over tre uger med fortsat opløsning af symptomer og forbedring af synsstyrken. Hun havde ingen gentagelse af hovedpine eller forværret syn under prednison-tilspidsningen. På hendes seneste udnævnelse, hun havde tilspidset ned til 5 mg hver anden dag, uden tilbagevenden af symptomer. Hendes synsstyrke ved det opfølgende besøg var 20/15 – 2 OD og 20/20+2 OS, og macular OCT viste fuld opløsning af skiveødem og serøse nethindeafdelinger i begge øjne (figur 4).
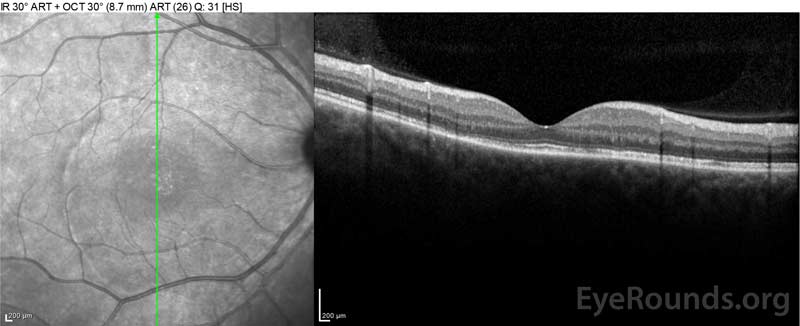
figur 4: Optisk kohærens-tomografi, der viser subretinalvæske ved baseline (øverst) og opløsningsforløbet efter en uge (midten) og fem uger (nederst), mens den er i en højdosis oral prednison-tilspidsning. Bemærk udjævningen ud af de choroidale bølger med behandling.
 |
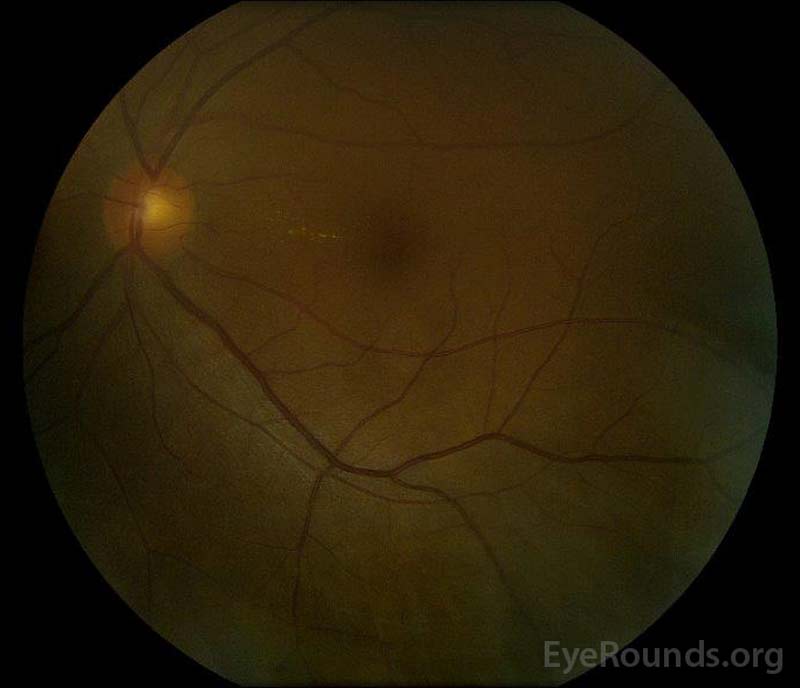 |
figur 5: Farvefundusfotografering af højre (a) og venstre (B) øjne under rekonvalescensfasen, der viser forbedring i subretinalvæsken og skiveødem.
diagnose
ufuldstændig Vogt-Koyanagi-Harada sygdom
diskussion
Vogt-Koyanagi-Harada (VKH) sygdom er en systemisk autoimmun tilstand, der er kendetegnet ved bilateral ikke-nekrotiserende granulomatøs panuveitis forbundet med ekstraokulære integumentære ændringer, såsom poliose og vitiligo, og betændelse, der påvirker uvea, indre øre, hår og meninges. Harada sygdom er den isolerede uveitis uden de systemiske tegn eller symptomer på VKH.
etiologi
etiologien af VKH-sygdom er stadig stort set ukendt på trods af den nuværende forskningsindsats. Det menes at være en erhvervet autoimmun sygdom, der involverer T-celle-medieret overfølsomhed over for melanocytiske selvantigener, med en underliggende genetisk disponering og mulig mikrobiel trigger . Tyrosinase og tyrosinase-relaterede peptider er melanocytantigener, der er blevet foreslået som mål for autoimmune processer i VKH . Imidlertid var øget risiko for VKH-sygdom ikke forbundet med tyrosinase-genfamilien, ifølge en undersøgelse .
på grund af den øgede forekomst blandt visse etniske grupper og kønsgrupper menes der at være en genetisk disponering i patogenesen af VKH. Flere gener, herunder humant leukocytantigen (HLA) og interleukin (IL) gener, har været forbundet med VKH i forskellige etniske populationer . HLA-receptorer er vigtige histokompatibilitetskomplekser hos mennesker, der præsenterer peptider til immunsystemet. HLA-DR1, HLA-DR4, HLA-DRB1*0405 og HLA-Drv53 er flere haplotyper, der findes hos patienter med VKH-sygdom . HLA-DR4 er mere almindelig hos japanske og spanske mennesker, mens HLA-DRB1*0405 er hyppigere hos koreanske og mellemøstlige patienter . Både HLA-DR4 og HLA-DRB1*0405 alleler findes hos vietnamesiske patienter . HLA-DRB1-receptoren binder til melanocytantigener i varierende kapacitet. På trods af disse foreninger anbefales genetisk testning ikke på nuværende tidspunkt.
i betragtning af de sædvanlige prodromale symptomer, der ledsager VKH, herunder feber, hovedpine, meningismus og tinnitus, er en tilskyndende viral etiologi blevet foreslået som en udløser for VKH-indtræden gennem mekanismer for molekylær efterligning hos genetisk disponerede patienter. Cytomegalovirus-konvolut glycoprotein H har signifikant aminosyrehomologi til tyrosinase-peptidet, og CMV-infektion kan udløse VKH gennem molekylær efterligning (dvs .genkendelse af HLA-klasse II-receptorer). Ebstein-bar virus (EBV) er også blevet impliceret. Der har imidlertid ikke været nogen endelige beviser for en viral etiologi af VKH, og det er fortsat uklart, hvad der udløser VKH autoimmun respons .
Patofysiologi
Der er fire klassiske faser af VKH, der kan have variable præsentationer: prodromal, akut uveitisk, rekonvalescent og kronisk tilbagevendende. Histopatologiske ændringer begynder typisk i den akutte fase .
den akutte uveitiske fase er kendetegnet ved bilateral uveal fortykning sekundært til granulomatøs betændelse. Granulomerne består af lymfocytter, makrofager og granulatfyldte epithelioide og gigantiske celler . Selvom epithelioidcellerne tidligere blev antaget at være ændrede melanocytter, foreslog en opfølgende immunhistokemisk undersøgelse i stedet en oprindelse fra vævsmakrofager . Granulomer fyldt med epithelioid histiocytter, betegnet Dalen-Fuchs knuder, kan ofte ses mellem retinal pigmentepitel (RPE) og Bruchs membran. Den uveal granulomatøse betændelse fører til choroidal fortykning og eksudative retinale løsrivelser fyldt med proteinholdig væske. Derudover pleocytose (i.e., øget celletal) kan være til stede i det forreste kammer og glaslegemet .
den rekonvalescerende fase identificeres ved depigmentering af choroid og ekstraokulære områder, herunder periokulær hud og hår. Et depigmenteret choroid sæt mod en bleg optisk nerve giver indtryk af en” solnedgang-glød ” fundus, som er et klassisk træk ved denne fase af VKH . Derudover bliver Dalen-Fuchs knuder mere fremtrædende under RPE i den rekonvalescerende fase .
den kronisk-tilbagevendende fase er kendetegnet ved nedsat choroidal tykkelse, opløsning af serøse nethindeafdelinger, kronisk mild vitritis og tilbagevendende granulomatøs betændelse i det forreste segment. Choroidal neovaskularisering (CNV) og subretinal fibrose kan udvikle sig i denne fase og er indikatorer for alvorlig sygdomsprogression . Katarakter og sekundær glaukom er andre komplikationer af langvarig eller tilbagevendende betændelse i denne fase .
Epidemiologi
VKH er udbredt i løb med mørkere hudpigment, især asiater, sydamerikanere, mellemøstlige og indianere. VKH sygdom tegner sig for > 10% af uveitis i disse populationer . Kun 1-4% af uveitis tilfælde menes at være sekundære til VKH sygdom i USA (7). I USA har de fleste tilfælde af VKH vist sig at påvirke individer af asiatisk, latinamerikansk og/eller indiansk anstændig . Interessant nok påvirker VKH-sygdom sjældent afrikanere på trods af deres mørke pigmentering . Forekomsten af VKH-sygdom varierer meget mellem racemæssige undergrupper i nabolandene . For eksempel er Koreas forekomst af VKH kun 2%, meget lavere end den, der findes i Japan og Kina .
VKH har en typisk begyndelse på 20 til 50 år ; undersøgelser antyder imidlertid, at 3,1-13,4% af VKH-tilfælde er pædiatriske patienter, og 10% af tilfældene er 65-årige . Klassisk, VKH menes at have en forkærlighed for det kvindelige køn, og mens de fleste undersøgelser viser, at VKH uforholdsmæssigt påvirker kvinder, et par undersøgelser har vist en mandlig disposition eller ingen kønsdisponering .
tegn/symptomer
som nævnt er de fire stadier af VKH-sygdom prodromal, uveitisk, rekonvalescent og kronisk tilbagevendende. Hvert trin udviser forskellige kliniske træk.
- Prodromal: denne indledende fase kan forekomme som en influensa-lignende sygdom med overvejende konstitutionelle symptomer, såsom hovedpine, svimmelhed, feber, træthed og / eller kvalme. Neurologiske symptomer på meningitis, kraniale nervepalsier og optisk neuritis samt auditive symptomer på tinnitus, dysacusis og svimmelhed er blevet rapporteret . Fotofobi, sløret syn, flydere og/eller øjensmerter begynder normalt inden for 48 timer efter prodromale symptomer . Den prodromale fase varer typisk fra et par dage til uger.
- akut Uveitisk: dette trin inkluderer sløret syn, fotofobi, konjunktival injektion og øjensmerter. Der kan være mild anterior uveitis, der i første omgang forekommer ikke-granulomatøs. Ensidig indtræden overgår typisk til bilateral involvering inden for 1-2 uger. Granulomatøs anterior uveitis med fårefedtkeratiske udfældninger kan udvikle sig. Posterior undersøgelsesresultater kan omfatte synsnerveødem og hyperæmi , multifokale områder af choroiditis, flere områder med serøse nethindeafdelinger lokaliseret til den bageste fundus, choroidal fortykning, udstrålende chorioretinale folder og vitritis . Serøse nethindeløsninger kan danne et kløverbladsmønster i den bageste fundus og kunne udvikle sig til omfattende bulløse løsrivelser i alvorlige tilfælde . Akut inflammatorisk glaukom har været forbundet med denne fase af sygdommen og kan præsentere med et lavt forkammer sekundært til ciliær kropsødem, der efterligner akut vinkellukning . Varigheden af den akutte uveitiske fase afhænger af den hurtige diagnose og styring.
- kronisk Uveitisk eller rekonvalescent: dette trin udvikler sig typisk flere uger efter den akutte fase og er kendetegnet ved vitiligo (f .eks. Depigmentering nær hornhinden limbus, kendt som Sugiuras tegn, kan ses en måned efter sygdomsdebut ; dette tegn ses dog sjældent uden for den japanske befolkning . Choroidal depigmentering forekommer normalt over et par måneder og resulterer i den lyse orange-røde farve på choroid og den klassiske “solnedgang glød fundus.”Sunset glød fundus menes at være den vigtigste og forudsigelige i diagnosen kronisk VKH . Veldefinerede, runde, nummulære chorioretinale ar kan dannes i midten af periferien. Den kroniske uveitiske fase varer typisk flere måneder.
- kronisk-tilbagevendende: Dette trin er kendetegnet ved tilbagevendende episoder af granulomatøs anterior uveitis med keratiske udfældninger af fårefedt, irisknuder, irisdepigmentering, posterior synechiae, posterior subkapsulær grå stær, sekundær glaukom, choroidale neovaskulære membraner og i sidste ende subretinal fibrose og nummulær chorioretinal atrofi . Den kroniske fase udvikler sig typisk mindst seks måneder efter den første præsentation. De serøse nethindeløsninger, der er til stede i de akutte og rekonvalescerende faser, gentager sig typisk ikke med aggressiv kortikosteroidbehandling .
diagnostiske kriterier
de seneste diagnostiske kriterier, der hedder de reviderede diagnostiske kriterier (RDC) for VKH, blev defineret i 1999 på det første internationale værksted om VKH . Disse er beskrevet i tabel 1. RDC er nyttige, idet de deler VKH i tre forskellige diagnostiske kategorier baseret på sygdomsfasen, hvor en patient præsenterer: komplet, ufuldstændig og sandsynlig. Denne kategorisering af sygdom muliggør passende og tidlig håndtering af “sandsynlig” sygdom, der kan hjælpe med at forhindre progression til “komplet” sygdom.
arbejde op for andre årsager til okulær inflammation, både infektiøs og auto-inflammatorisk, er afgørende. Disse kan omfatte erythrocytsedimenteringshastighed (ESR), C-reaktivt protein (CRP), kvantiferon-guld test for tuberkulose, hurtig plasma reagin (RPR) for syfilis, angiotensin-konvertering (ACE) og en røntgenbillede af brystet for sarkoidose, antinukleært antistof (ANA) og p-/c-ANCA. Også en historie med nyligt okulært traume eller intraokulær kirurgi skal bemærkes og antyder sandsynligvis sympatisk ophthalmia (SO) som den mere sandsynlige diagnose givet den meget lignende præsentation og patofysiologi delt mellem SO og VKH .
for at understøtte en diagnose af VKH i tvetydige tilfælde kan en lumbal punktering udføres for at se efter lymfocytisk og monocytisk pleocytose; dette anvendes dog sjældent klinisk. Otteogtreds procent af patienterne har pleocytose i cerebrospinalvæsken (CSF) inden for en uge, og 97% har pleocytose inden for tre uger. Forhøjede niveauer af immunceller kan vare op til otte uger efter sygdomsdebut . T-celleoverflademarkørprofilerne er ens mellem CSF og den vandige humor, men adskiller sig fra blodet. Dette antyder CSF ‘ s evne til nøjagtigt at afspejle uveal inflammation i VKH sygdom .
tabel 1. Reviderede diagnostiske kriterier for Vogt-Koyanagi-Harada sygdom
*fra tabel 1 in (15).
“komplet Vogt-Koyanagi-Harada sygdom (kriterier 1 til 5 skal være til stede)
- ingen historie med gennemtrængende okulært traume eller kirurgi forud for den første begyndelse af uveitis.
- ingen kliniske eller laboratorie beviser, der tyder på andre okulære sygdomsenheder. Bilateral okulær involvering (A eller b skal være opfyldt afhængigt af sygdomsstadiet, når patienten undersøges).
- tidlige manifestationer af sygdom.
- der skal være tegn på en diffus choroiditis (med eller uden anterior uveitis, glasagtig inflammatorisk reaktion eller optisk diskhyperæmi), som kan manifestere sig som et af følgende:
- tidlige manifestationer af sygdom.
- fokale områder af subretinalvæske eller
- Bullous serous retinal detachments.
- med tvetydige fundusfund; begge følgende skal også være til stede:
- fokale områder med forsinkelse i choroidal perfusion, multifokale områder med lokalisere lækage, store placoide områder med hyperfluorescens, pooling i subretinalvæske og optisk nervefarvning (opført i rækkefølge efter sekventielt udseende) ved fluoresceinangiografi og
- diffus choroidal fortykning uden tegn på posterior skleritis ved ultralyd.
- sene manifestationer af sygdom.
- historie, der tyder på tidligere tilstedeværelse af fund fra 3a, og enten begge (2) og (3) nedenfor eller flere tegn fra (3):
- okulær depigmentering (en af følgende manifestationer er tilstrækkelig): (a) solnedgang glød fundus, eller (b) Sugiura tegn.
- andre okulære tegn:
- Nummular chorioretinal depigmented ar, eller
- retinal pigmentepitel klumpning og/eller migration, eller
- tilbagevendende eller kronisk anterior uveitis.
- neurologiske / auditive fund (kan være løst efter undersøgelsestidspunktet).
- Meningismus (utilpashed, feber, hovedpine, kvalme, mavesmerter, stivhed i nakke og ryg eller en kombination af disse faktorer; hovedpine alene er imidlertid ikke tilstrækkelig til at opfylde definitionen af meningismus) eller
- Tinnitus eller
- Cerebrospinalvæskepleocytose.
- Integumentary fund (ikke forud for begyndelsen af centralnervesystemet eller okulær sygdom).
- alopeci, eller
- Poliosis, eller
- vitiligo.
ufuldstændig Vogt-Koyanagi-Harada-sygdom (kriterium 1 til 3 og enten 4 eller 5 skal være til stede)
- ingen historie med gennemtrængende okulært traume eller kirurgi forud for den første begyndelse af uveitis, og
- ingen kliniske eller laboratoriebeviser, der tyder på andre okulære sygdomsenheder, og
- Bilateral okulær involvering.
- neurologiske/auditive fund; som defineret for komplet Vogt-Koyanagi-Harada sygdom ovenfor eller
- integumentære fund; som defineret for komplet Vogt-Koyanagi-Harada sygdom ovenfor.
sandsynlig Vogt-Koyanagi-Harada sygdom (isoleret okulær sygdom; kriterier 1 til 3 skal være til stede)
- ingen historie med gennemtrængende okulært traume eller kirurgi forud for den første begyndelse af uveitis.
- ingen kliniske eller laboratorie beviser, der tyder på andre okulære sygdomsenheder.Bilateral okulær involvering som defineret for komplet Vogt-Koyanagi-Harada sygdom ovenfor. “
test/Laboratorieoparbejdning
i den første oparbejdning af VKH bør man overveje at opnå følgende tests:
- optisk kohærens tomografi (OCT): I den akutte uveitiske fase vil OCT sandsynligvis vise signifikant koroidfortykning og serøse nethindeløsninger. De subretinale væskeakkumuleringer kan have septationer, der antages at være fibrinmembraner og inflammatoriske produkter, hvilket skaber en lobulær struktur, der også kan ses på fluoresceinangiografi. I rekonvalescensfasen kan OCT detektere områder med nethindefortynding efter løst betændelse efter kortikosteroidbehandling .
- B-scan ultralyd: I den akutte fase kan ultrasonografi vise diffus posterior choroidal fortykning, posterior skleral fortykning, nethindeafskillelser og glasagtige opaciteter . Ciliære effusioner kan observeres med ultralydbiomikroskopi . Denne test er også nyttig til at udelukke posterior skleritis. fluoresceinangiografi (FA): klassisk afslører FA multifokale choroidale hypofluorescerende prikker i den tidlige fase efterfulgt af flere fokale hyperfluorescerende områder med diffus lækage i den sene fase . Farvestoffet lækker gennem RPE og akkumuleres i det subretinale rum, der omgiver de hyperfluorescerende prikker. FA kan være diagnostisk nyttigt, når VKH sygdom præsenterer uden ekstraokulære symptomer. Optisk diskhyperfluorescens og vinduesdefekter forårsaget af atrofiske chorioretinale ar kan ses i midten af periferien . FA i det kronisk tilbagevendende stadium af VKH-sygdom viser uspecifikke vinduesdefekter på grund af RPE-skade, choroidal neovaskularisering og subretinal fibrose .
- indocyanin grøn (ICG) angiografi: Tidlig fase ICG skildrer hyperfluorescerende stromale kar, der indikerer choroidal vaskulopati og hypofluorescerende mørke prikker, der svarer til granulomer og forsinket ujævn fyldning af choroidal vaskulatur . Den sene fase afslører uklar stromale vaskulære mønstre og diffus choroidal hyperfluorescens. Diskhyperfluorescens antyder alvorlig sygdom. ICGA kan detektere subklinisk choroidal inflammation i meget tidlige stadier eller endda efter systemisk terapi .
- lumbalpunktur: pleocytose i cerebrospinalvæsken er til stede hos de fleste VKH-patienter. Lumbalpunktur skal udføres tidligt i sygdomsforløbet, da pleocytose kan løse
behandling/Ledelse/retningslinjer
behandlingsmål i VKH inkluderer tidlig diagnose og undertrykkelse af aktiv betændelse sammen med forebyggelse af tilbagevendende betændelse og synstruende komplikationer, såsom glaukom, bulløs nethindeløsning og choroid neovaskularisering.
systemisk kortikosteroidbehandling er den foretrukne behandling for VKH-sygdom, især i det akutte uveitiske Stadium. Det har vist sig, at indgivelsesvejen for kortikosteroid (oral versus intravenøs) ikke påvirker synsstyrken eller forekomsten af visuelt signifikante komplikationer ved behandling af akut VKH . Ved alvorlig sygdom er den foreslåede protokol intravenøs administration af methylprednisolon i tre dage efterfulgt af oral behandling med høj dosis prednison. Ved mild-moderat sygdom kan højdosis oral prednison være tilstrækkelig ved 1-2 mg/kg/dag. Steroiddosis bør langsomt tilspidses over ca. seks måneder for at forhindre gentagelse . Aggressiv tidlig behandling sammen med seriel FA-test, der viser forsvinden af farvestoflækage gennem RPE, kan hjælpe med at forhindre yderligere sygdomsprogression, gentagelse og ekstraokulære manifestationer . Aktuelle steroider og cycloplegics kan reducere celler i det forreste kammer og glasagtige humor.
Intravitreal og sub-Tenon injektioner af triamcinolon er blevet anvendt til kortvarig kontrol af intraokulær inflammation i de akutte eller tilbagevendende faser; disse lokale terapier bør overvejes i tilfælde af recalcitrant sygdom og hos patienter, der dårligt tolererer de ugunstige systemiske bivirkninger af steroider i betragtning af den udvidede steroidtilspidsning. Intravitreal anti-VEGF injektioner anvendes undertiden til kontrol af choroidal neovaskularisering og i tilfælde af vedvarende foveal serøse retinale løsrivelser .
Steroidsparende midler inklusive antimetabolitter, calcineurinhæmmere, biologics, TNF-alfa-hæmmere eller cytotoksiske midler kan bruges til behandling af VKH og bør overvåges nøje, ofte i koordinering med en reumatologitjeneste . Der har været løbende diskussion om brugen af ikke-steroide immunsuppressive midler som førstelinjebehandling for VKH-sygdom. En nylig undersøgelse afslørede imidlertid ingen forskelle i resultater mellem tidlig førstelinjeimmunmodulerende behandling (IMT) og prednisonbehandling alene . Desuden er immunsuppressive og biologiske terapier dyre og kræver omhyggelig evaluering forud for behandlingen samt hyppig opfølgning med blodarbejde for at vurdere for alvorlige bivirkninger.
i det kronisk tilbagevendende stadium kan hyppig gentagelse antyde resistens over for kortikosteroidbehandling og antyder behov for steroidbesparende immunmodulerende behandling . Det foretrukne middel til steroidresistent gentagelse eller steroidintolerance er cyclosporin . Interferon alfa-2a er biologiske midler, der også er blevet anvendt til behandling af refraktær uveitis ved VKH-sygdom.
for at behandle den forreste uveitis, der ofte er forbundet med akut VKH, skal topiske steroider (f.eks. prednisolonacetat 1%) og topisk cyloplegi (f. eks. cyclopentolat 1% eller atropin 1%) ordineres afhængigt af graden af betændelse i det forreste kammer.
okulære komplikationer er ofte forbundet med VKH-sygdom. I betragtning af de mange faser og forskellige præsentationer, hvor en patient kan præsentere med VKH, kan behandlingen i mange tilfælde forsinkes. I alvorlige former for VKH og i gentagelser kan intraokulær betændelse være vanskelig at kontrollere og kan resultere i strukturel skade. Over 50% af patienterne udvikler relaterede komplikationer, herunder grå stær, sekundær glaukom, choroidale neovaskulære membraner, subretinal fibrose eller en kombination af disse (figur 6) .
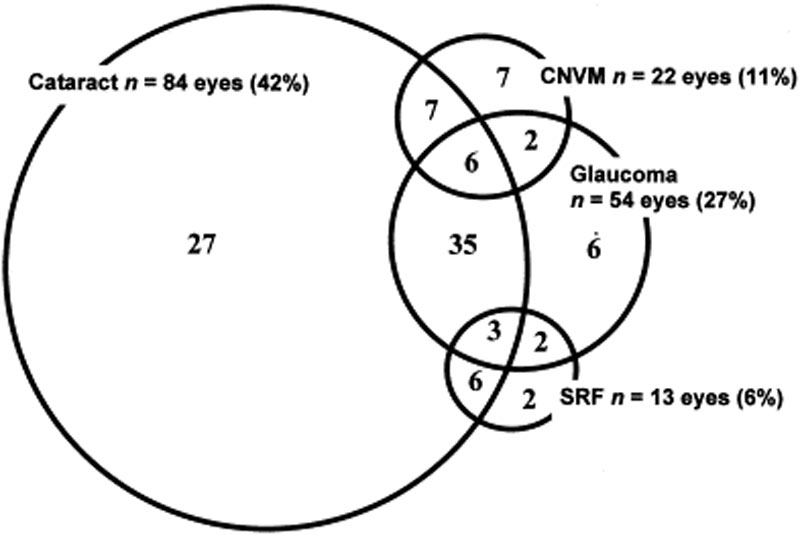
figur 6: Venn-diagram, der viser komplikationer hos VKH-patienter. (Bruges med tilladelse fra Am J Ophthalmol. 2001;131(5):599-606 )
epidemiologi og etiologi
|
SIGNS
|
symptomer
|
behandling/behandling
|
- Du L, Kijlstra A, Yang P. Vogt-Koyanagi-Harada sygdom: ny indsigt i patofysiologi, diagnose og behandling. Prog Retin Øje Res 2016; 52:84-111. https://PubMed.gov/26875727. DOI: 10.1016 / j. preteyeres.2016.02.002
- Yamaki K, Gocho K, Hayakava K, Kondo I, Sakuragi S. Tyrosinase-familieproteiner er antigener, der er specifikke for Vogt-Koyanagi-Harada-sygdom. J Immunol 2000;165(12):7323-7329. https://PubMed.gov/11120868
- Horie Y, Takemoto Y, miyasaki A, Namba K, Kase s, Yoshida K, Ota M, Hasumi Y, Inoko H, Miuki N, Ohno S. Tyrosinase genfamilie og Vogt-Koyanagi-Harada sygdom hos japanske patienter. Mol Vis 2006; 12: 1601-1605. https://PubMed.gov/17200659
- Ng JY, Luk FO, Lai TY, Pang CP. Indflydelse af molekylær genetik i Vogt-Koyanagi-Harada sygdom. J Oftalmisk Inflamm Inficere 2014; 4: 20. https://PubMed.gov/25097674. DOI: 10.1186 / s12348-014-0020-1 B. Uveitis. Kanskis kliniske oftalmologi, Ny York: Elsevier; 2016; kapitel 11; s.395-465.
- Yeh PT YC, Yang CH, Lin CP. Nonrhegmatogen Retinal Detachment. I: Schachat AP SS, Hinton DR, Vilkinson CP, Viedemann P,, Redaktør. Ryans nethinde. Ny York: Elsevier; 2018; kapitel 99; s.1828-1849.
- Goto H RK, Rao N. Vogt-Koyanagi-Harada sygdom. I: Schachat AP SS, Hinton DR, Vilkinson CP, Viedemann P, Redaktør. Ryans nethinde. Ny York, Ny York: Elsevier; 2018; kapitel 78; s.1505-1515.
- Riddington L, Hall AJ, Tait B, Nicholson I, Varney M. Vogt-Koyanagi-Harada syndrom hos patienter af vietnamesisk herkomst. Aust N J Ophthalmol 1996; 24 (2): 147-149. https://PubMed.gov/9199747
- Sugita S, Takase H, Takuchi T, Taguchi C, Mochisuki M. Krydsreaktion mellem tyrosinase peptider og cytomegalovirus antigen af T-celler fra patienter med Vogt-Koyanagi-Harada sygdom. Int Ophthalmol 2007; 27(2-3):87-95. https://PubMed.gov/17253112. DOI: 10.1007 / s10792-006-9020-Y
- Freund BK SD, MIELER VF, Yannon LA. Betændelse. Retinal Atlas. Ny York, Ny York: Elsevier 2017; kapitel 4; s.279-398.
- Rao N. Vogt-Koyanagi-Harada sygdom. I: J YMaD, redaktør. Oftalmologi. Ny York, Ny York: Elsevier; 2014; kapitel 7.17; s.761-763.
- Rao NA, s, Font RL. Sympatisk ophthalmia. En immunhistokemisk undersøgelse af epithelioid og gigantiske celler. Oftalmologi 1985; 92(12):1660-1662. https://PubMed.gov/4088616
- Nussenblatt RB. Vogt-Koyanagi-Harada Syndrom. I: hvidkål RBNaSM, redaktør. Uveitis: grundlæggende og klinisk praksis. 4. udgave ed: Elsevier; 2010; kapitel Kapitel 24.
- Læs RV, Holland GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L, Pivetti-P, Tessler HH, Usui M. reviderede diagnostiske kriterier for Vogt-Koyanagi-Harada sygdom: rapport fra et internationalt udvalg for nomenklatur. Am J Ophthalmol 2001; 131(5):647-652. https://PubMed.gov/11336942
- Chung H, Choi GD. Klinisk analyse af uveitis. Koreansk J Ophthalmol 1989; 3(1):33-37. https://PubMed.gov/2795939. DOI: 10.3341 / kjo.1989.3.1.33
- Abu El-Asrar AM, al-Kharashi AS, Aldibhi H, Al-Fraykh H, Kangave D. Vogt-Koyanagi-Harada sygdom hos børn. Øje (Lond) 2008;22(9):1124-1131. https://PubMed.gov/17479116. DOI: 10.1038 / sj.øjne.6702859
- Martin TD, Rathinam SR, Cunningham et. Prævalens, kliniske egenskaber og årsager til synstab hos børn med Vogt-Koyanagi-Harada sygdom i Sydindien. Nethinden 2010; 30(7):1113-1121. https://PubMed.gov/20168275. DOI: 10.1097 / IAE.0b013e3181c96a87
- Forster DJ, grøn RL, Rao NA. Unilateral manifestation af Vogt-Koyanagi-Harada syndrom hos et 7-årigt barn. Am J Ophthalmol 1991; 111 (3): 380-382. https://PubMed.gov/2000916
- Yamamoto y, Fukushima A, Nishino K, Koura Y, Komatsu T, Ueno H. Vogt-koyanagi-Harada sygdom med debut hos ældre patienter i alderen 68 til 89 år. Jpn J Ophthalmol 2007; 51 (1): 60-63. https://PubMed.gov/17295144. DOI: 10.1007 / s10384-006-0379-0
- Vang y, Chan CC. Kønsforskelle i vogt-koyanagi-Harada sygdom og sympatisk ophthalmia. J Ophthalmol 2014; 2014: 157803. https://PubMed.gov/24734166. DOI: 10.1155/2014/157803 Nakao K, Abematsu N, Misushima Y, Sakamoto T. Optic disc hævelse i Vogt-Koyanagi-Harada sygdom. Invest Ophthalmol Vis Sci 2012; 53 (4): 1917-1922. https://PubMed.gov/22408010. DOI: 10.1167 / iovs.11-8984
- Rao NA, Gupta A, Dustin L, Chee SP, Okada AA, Khairallah M, Bodaghi B, Lehoang P, Accorinti M, Mochisuki M, Prabriputaloong T, Læs RV. Hyppigheden af at skelne kliniske træk i Vogt-Koyanagi-Harada sygdom. Ophthalmology 2010;117(3):591-599, 599.e591. https://PubMed.gov/20036008. DOI: 10.1016/j.ophtha.2009.08.030
- Veerappan M, Fleischman D, Ulrich JN, Stinnett SS, Jaffe GJ, Allingham RR. The Relationship of Vogt-Koyanagi-Harada Syndrome to Ocular Hypertension and Glaucoma. Ocul Immunol Inflamm 2017;25(6):748-752. https://PubMed.gov/27438521. DOI: 10.1080/09273948.2016.1189578
- Baltmr A, Lightman S, Tomkins-Netzer O. Vogt-Koyanagi-Harada syndrome – current perspectives. Clin Ophthalmol 2016;10:2345-2361. https://PubMed.gov/27932857. DOI: 10.2147/OPTH.S94866
- Kitaichi N, Matoba H, Ohno S. Den positive rolle lumbal punktering i diagnosen Vogt-Koyanagi-Harada sygdom: lymfocyt delmængder i vandig humor og cerebrospinalvæske. Int Ophthalmol 2007; 27(2-3):97-103. https://PubMed.gov/17211585. DOI: 10.1007 / s10792-006-9016-7
- Oshima Y, Harino s, Hara Y, Tano Y. indocyaningrønne angiografiske fund i Vogt-Koyanagi-Harada sygdom. Am J Ophthalmol 1996; 122 (1): 58-66. https://PubMed.gov/8659599
- Læs RV, Yu F, Accorinti M, Bodaghi B, Chee SP, Fardeau C, Goto h, Holland GN, kV h, Kojima E, Lehoang P, Lemaitre C, Okada AA, Pivetti-P, Secchi a, se RF, Tabbara KF, Usui M, Rao NA. Evaluering af effekten på resultaterne af administrationsvejen for kortikosteroider ved akut Vogt-Koyanagi-Harada sygdom. Am J Ophthalmol 2006; 142(1):119-124. https://PubMed.gov/16815259. DOI: 10.1016 / j. ajo.2006.02.049
- Rubsamen PE, Gass JD. Vogt-Koyanagi-Harada syndrom. Klinisk kursus, terapi og langsigtet visuelt resultat. Arch Ophthalmol 1991;109(5):682-687. https://PubMed.gov/2025171
- Ursua CA, Velaska V, Sabat P, Berger O, Ramires S, Goecke A, V, Gatica H, Guerrero J. tidligere immunmodulerende behandling er forbundet med bedre visuelle resultater i en delmængde af patienter med Vogt-Koyanagi-Harada sygdom. Acta Ophthalmol 2015; 93 (6): e475-480. https://PubMed.gov/25565265. DOI: 10.1111 / aos.12648
- Læs RV, Rechodouni A, Butani N, Johnston R, LaBree LD, Smith RE, Rao NA. Komplikationer og prognostiske faktorer i Vogt-Koyanagi-Harada sygdom. Am J Ophthalmol 2001; 131(5):599-606. https://PubMed.gov/11336934
foreslået Citationsformat
Mai AP, Tran C, Vilson, ræv AR, Boldt HC. Vogt-Koyanagi-Harada (VKH) sygdom. EyeRounds.org.1. April 2019. Tilgængelig fra http://EyeRounds.org/cases/284-vogt-koyanagi-harada.htm
Leave a Reply