Vogt-Koyanagi-Harada (VKH) Malattia
Autori: Anthony P. Mai, BS; Charlene Tran, BS; Caroline W. Wilson, MD; Austin R. Fox, MD; H. Culver Boldt, MD
1 aprile 2019
diagnosi
Denuncia Capo
visione Sfocata e mal di testa
Storia del Presente Malattia
44-year-old donna Vietnamita presentato al dipartimento di emergenza con 10 giorni di storia della progressiva sfocata visione in entrambi gli occhi e un tre giorni di storia di forti mal di testa. La sua perdita della vista centrale non era migliorata con una rifrazione dal suo optometrista. I suoi gravi mal di testa occipitali peggiorarono con il movimento e furono associati a malessere generalizzato, estrema stanchezza, lieve fotofobia e lacrimazione. Il paracetamolo ha parzialmente alleviato il dolore.
Aveva recentemente viaggiato in Vietnam, ma ha negato di incontrare contatti malati lì. Ha negato claudicatio mascella, febbri, o variazioni di peso. Ha negato eruzioni cutanee, cambiamenti dell’udito, tinnito, vertigini, intorpidimento o formicolio. Ha negato di aver mai avuto la tubercolosi. Non aveva precedenti di problemi alla vista, malattie autoimmuni o cancro.
Passato Oculare Storia
- Storia dell’estetica chirurgia delle palpebre (bilaterale blefaroplastica) tre anni prima
- No storia dell’oculare trauma o malattia
Storia Medica
Nessuno
Farmaci
Paracetamolo come necessario
Allergie
Non noto farmaco allergie
Storia di Famiglia
Nessuna storia di malattia dell’occhio o malattia autoimmune
Storia Sociale
Lei, immigrati dal Vietnam diversi anni prima della presentazione. È sposata e ha tre figli. Lavora in un salone di bellezza. Non consuma prodotti del tabacco, alcol o sostanze illecite. Si reca in Vietnam ogni sei-dodici mesi.
Revisione
Negativo ad eccezione di quanto descritto la storia del presente malattia
OCULARE ESAME
l’Acuità Visiva con/senza correzione (Snellen)
- occhio Destro (OD): 20/300 (nessun miglioramento con il foro stenopeico)
- occhio Sinistro (OS): 20/60-2+2 (nessun miglioramento con il foro stenopeico)
Motilità Oculare/Allineamento
Completa extraoculari movimenti in entrambi gli occhi (OU)
la Pressione Intraoculare (IOP): (Tonopen)
- OD: 12 mmHg
- OS: 14 mmHg
Studenti
- OD: 4 mm scuro, 3 mm in luce, non relativo difetto pupillare afferente (RAPD)
- OS: 4 mm scuro, 3 mm di luce, non RAPD
Confronto visivo campi: (Contare con le dita)
- OD: scotoma Centrale
- OS: Totale inferotemporal difetto
Esterni
Normale su entrambi i lati
lampada a Fessura esame
- Coperchi/ciglia: Normale OU
- Congiuntiva/sclera: Chiara e tranquilla OU
- Cornea: 1+ punctata erosioni epiteliali, non keratic precipita OU
- camera Anteriore: Traccia delle cellule e flare e profondo OU
- Iris: Normale architettura OU
- Lente: Chiaro OU
Dilatato l’esame (DFE)
- Vitreo: Traccia vitreo anteriore cellule OU
- Disco:
- OD: Grado 3 disco edema, iperemica
- OS: Grado 2-3 disco edema, iperemica
- Coppa-di-disco rapporto: 0.0 OU
- Macula:
- OD: 3+ cystoid edema maculare (CME) e fluido sottoretinico (SRF) che si estende dal disco temporale macula. Senza lipidi o essudati. Coroide che appare paludosa.
- OS: 2 + CME e SRF che si estendono dal disco attraverso la fovea. 1-2 + lipide lineare che si estende dal disco verso la fovea. Coroide che appare paludosa.
- Vasi:
- OD: Guaina temporalmente
- OS: Normale
- Periferia:
- OD: Cistica della retina ciuffo anteriore all’equatore alle 10:30
- OS: Bassa SRF anteriore all’equatore a 4:00
 |
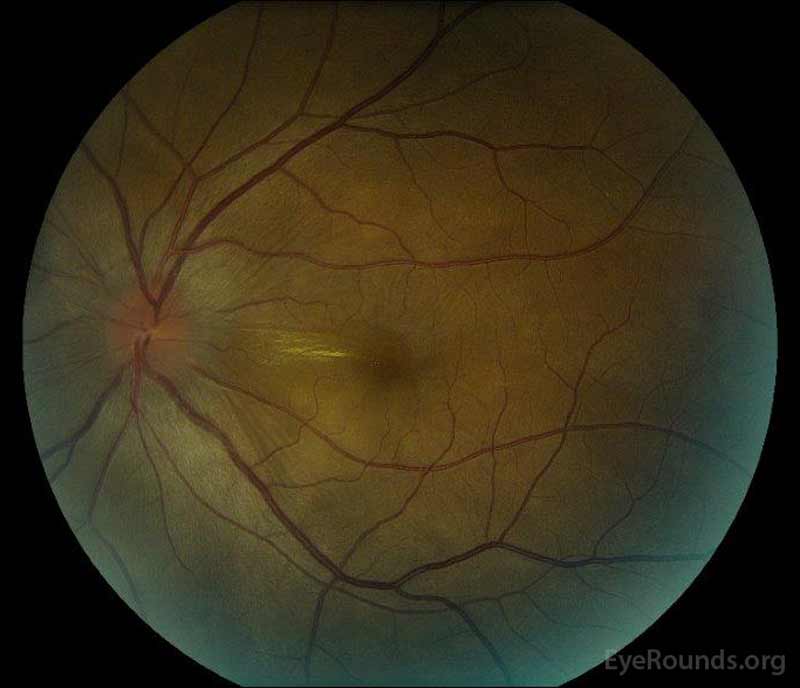 |
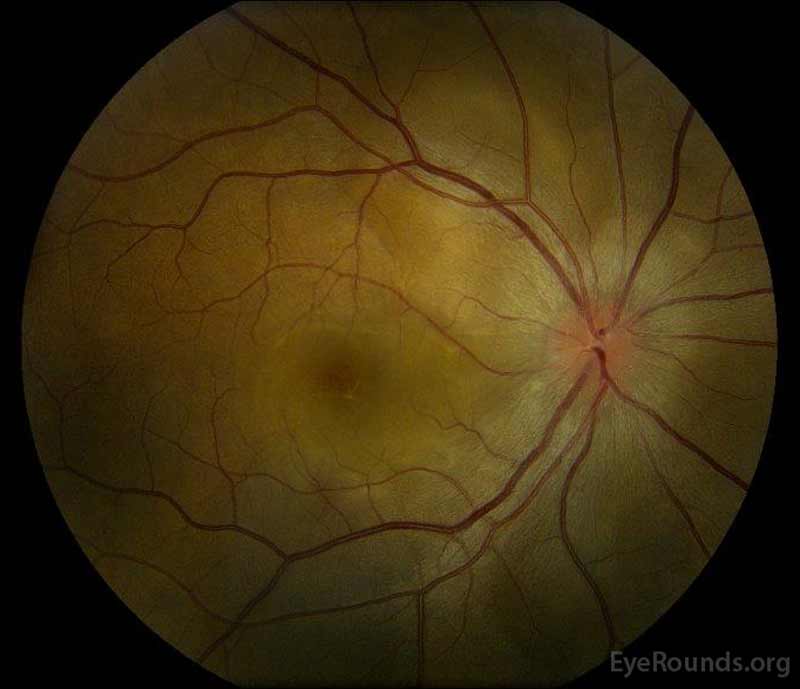
Figura 1: il Colore di fondo di fotografie alla presentazione: (immagine a Sinistra) L’occhio destro, disco edema e lieve iperemia nonché al fluido che si estende dal disco temporalmente attraverso la macula. C’è anche un distacco retinico sieroso focale superotemporale al disco, lungo il porticato superiore. (Right image) The left eye has disc edema and mild hyperemia, along with subretinal fluid extending from the disc to the macula and linear lipid deposits in the nasal macula.
 |
 |
 |
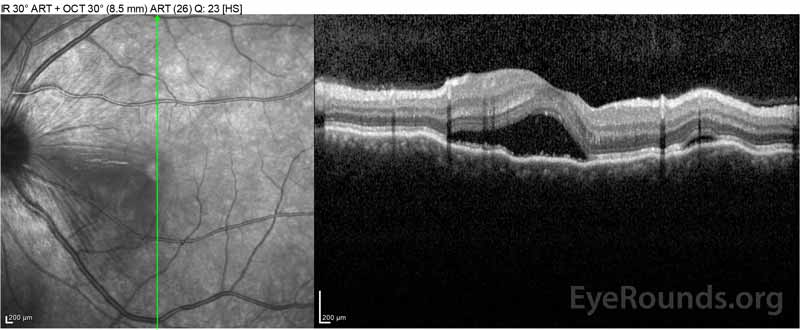 |
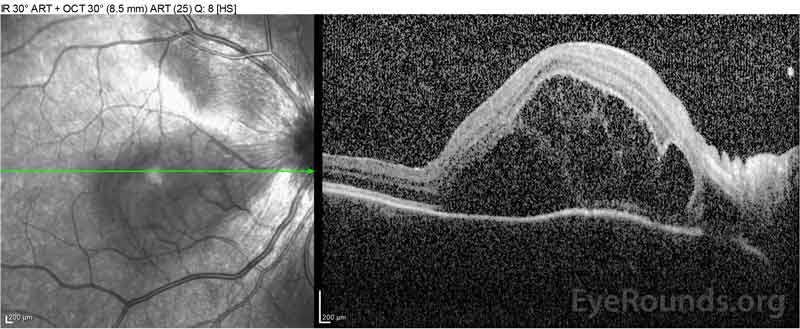
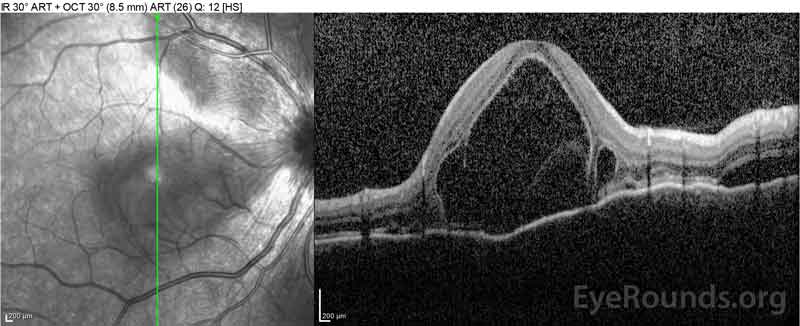
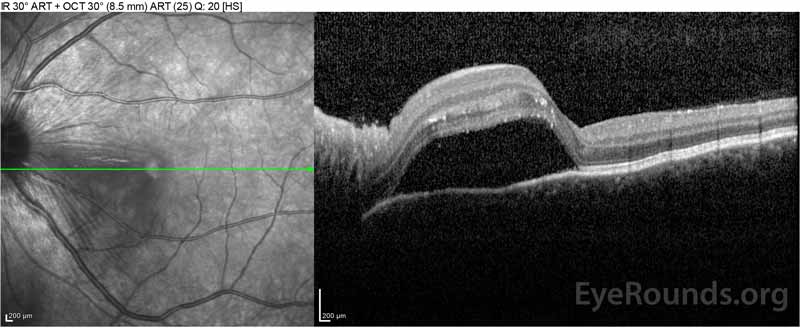
Figure 2: La tomografia a coerenza ottica (OCT) dell’occhio destro (pannelli superiori) mostra un distacco sieroso della retina che coinvolge la fovea con ampio liquido intraretinico sovrastante, interruzione degli strati retinici esterni e ondulazioni della coroide ispessita. OCT dell’occhio sinistro (pannelli inferiori) mostra un distacco sieroso della retina nella macula nasale che si estende fino alla fovea.
 |
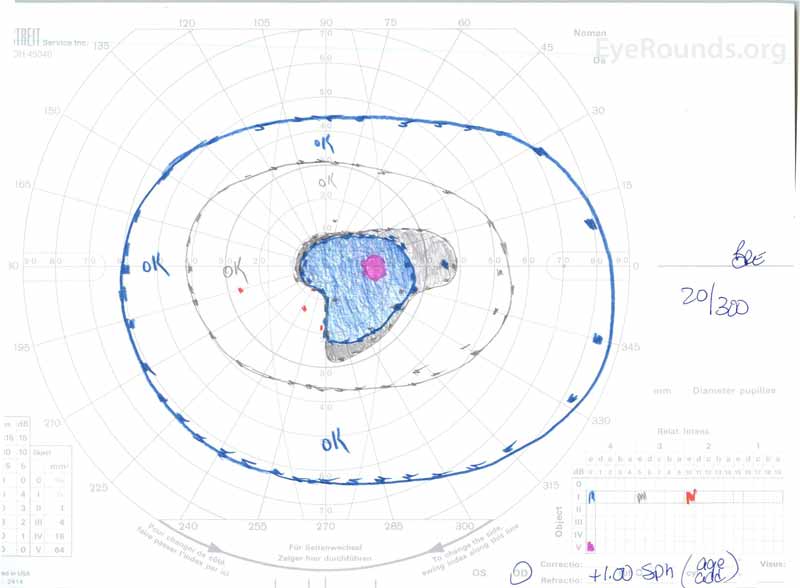 |
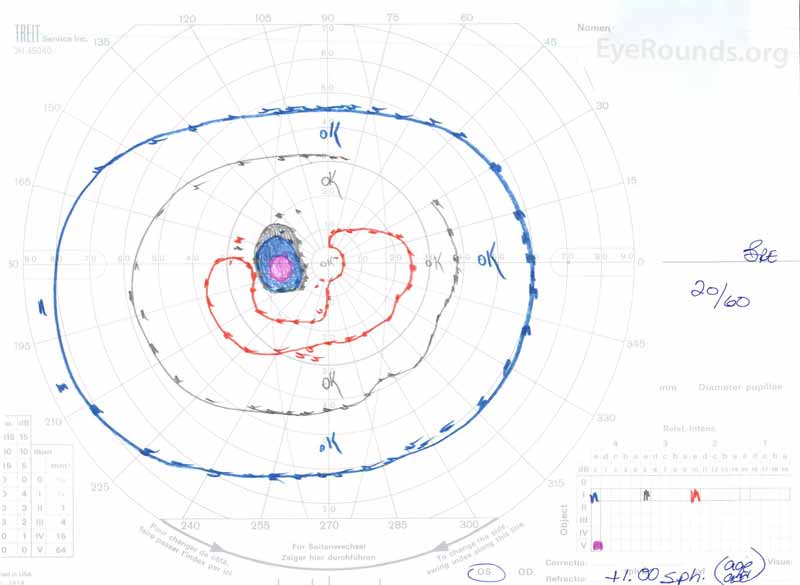
Figura 3: Goldman visual fields( GVF), OU. (Immagine a sinistra) OS mostra un punto cieco fisiologico ingrandito e un lieve scotoma centrale. (Immagine a destra) OD mostra uno scotoma centrale moderatamente denso che incorpora il punto cieco fisiologico e si estende inferotemporalmente.
B-scan: No segni di scleritis, lieve opacità vitreali/cellule inferiormente
Diagnosi Differenziale
- Acuta posteriore multifocale placoid pigmento epitheliopathy (APMPPE)
- sierosa Centrale chorioretinopathy
- la neurite Ottica
- Panuveitis
- malattie Autoimmuni (ad esempio, SLE, sarcoidosi)
- Infezione (e.g., la sifilide, la tubercolosi, la Bartonella henselae)
- Malignità (ad esempio, linfoma oculare)
- Posteriore scleritis
- oftalmia Simpatica
- Uveale effusione sindrome
- Vogt-Koyanagi-Harada Sindrome
WORK-UP
conteggio Completo del sangue
numero di globuli Bianchi: 4.9 K/mm3 (Rif: 3.7-10.5)
numero di globuli Rossi 3.99 M/mm3 (Rif: 4.0-5.2)
Emoglobina 11,6 g/dL (Rif: 11.9-15.5)
Ematocrito 35 % (Rif: 35-47)
Basic metabolic panel
Sodium 138 mEq/L (Ref: 135-145)
Potassium 4.3 mEq/L (Ref: 3.5-5.0)
Chloride 107 mEq/L (Ref: 95-107)
CO2 20 mEq/L (Ref: 22-29)
Blood urea nitrogen 16 mEq/dL (Ref: 10-20)
Creatinine 0.7 mg/dL (Ref: 0.5-1.0)
C-reactive Protein (CRP): <0.5 mg/dL (Ref: <=0.5)
Erythrocyte sedimentation rate (ESR): 12 mm/Hr (Ref: 0-20)
Angiotensin–converting enzyme (ACE): 13 U/L (Ref: 8-52)
QuantiFERON-TB Gold: negativo
Ferro, sangue 54 microgrammi / dL (Ref: 37-145)
Capacità totale di legame del ferro 379 microgrammi/dL (Ref: 250-425)
DECORSO CLINICO
Il paziente è stato inizialmente valutato dal dipartimento di emergenza a causa delle sue lamentele di mal di testa e perdita della vista. La tomografia computerizzata cerebrale (CT) e la risonanza magnetica (MRI) erano insignificanti. VES e CRP erano entro i livelli normali. La clinica oftalmologica l’ha valutata il giorno seguente e ha trovato distacchi sierici della retina bilaterali e panuveite. I laboratori ACE e QuantiFERON-TB Gold erano entrambi negativi. Le è stata diagnosticata la malattia di Vogt-Koyanagi-Harada in base alla sua presentazione clinica e alla sua discendenza asiatica. È stata trattata con 80 mg di prednisone al giorno, paracetamolo come necessario per il mal di testa, e vitamina D e supplementazione di calcio. I suoi mal di testa si risolsero rapidamente e la sua acuità visiva migliorò costantemente nelle due settimane successive. Il suo dosaggio di prednisone è stato poi ridotto a 40 mg in tre settimane con una risoluzione continua dei sintomi e un miglioramento dell’acuità visiva. Non ha avuto recidive di mal di testa o peggioramento della vista durante il cono del prednisone. Al suo appuntamento più recente, si era ridotta a 5 mg a giorni alterni, senza ritorno dei sintomi. La sua acuità visiva a quella visita di follow-up era 20/15-2 OD e 20/20+2 OS, e OCT maculare ha mostrato piena risoluzione di edema del disco e distacchi sierici della retina in entrambi gli occhi (Figura 4).
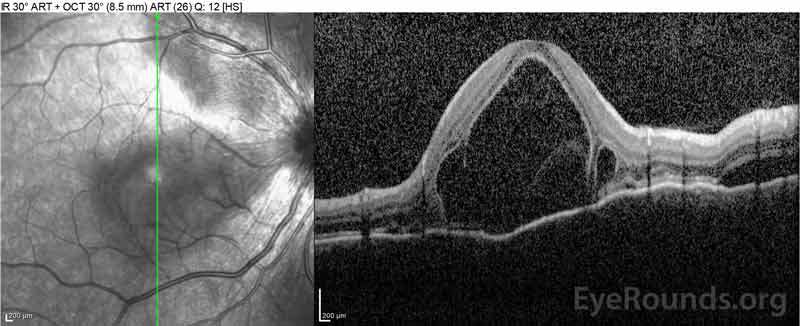
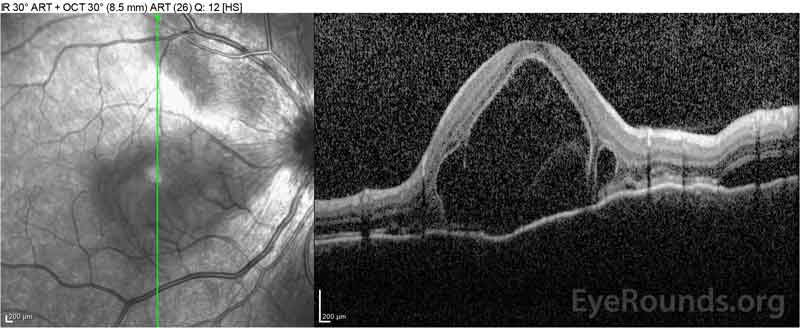
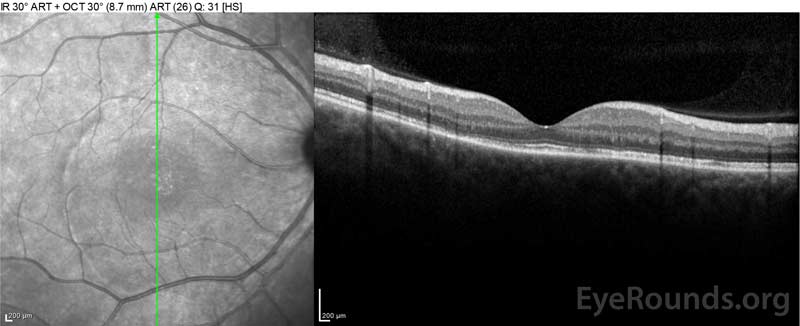
Figura 4: Tomografia a coerenza ottica che mostra il liquido subretinico al basale (in alto) e il decorso della risoluzione a una settimana (al centro) e a cinque settimane (in basso) mentre si è trattati con un cono di prednisone orale ad alte dosi. Si noti la levigatura delle ondulazioni coroidali con il trattamento.
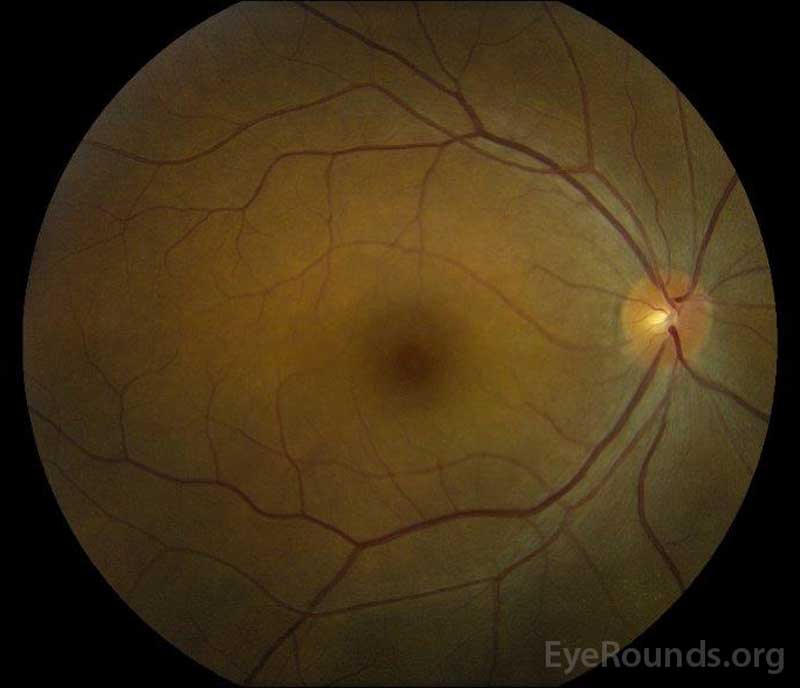 |
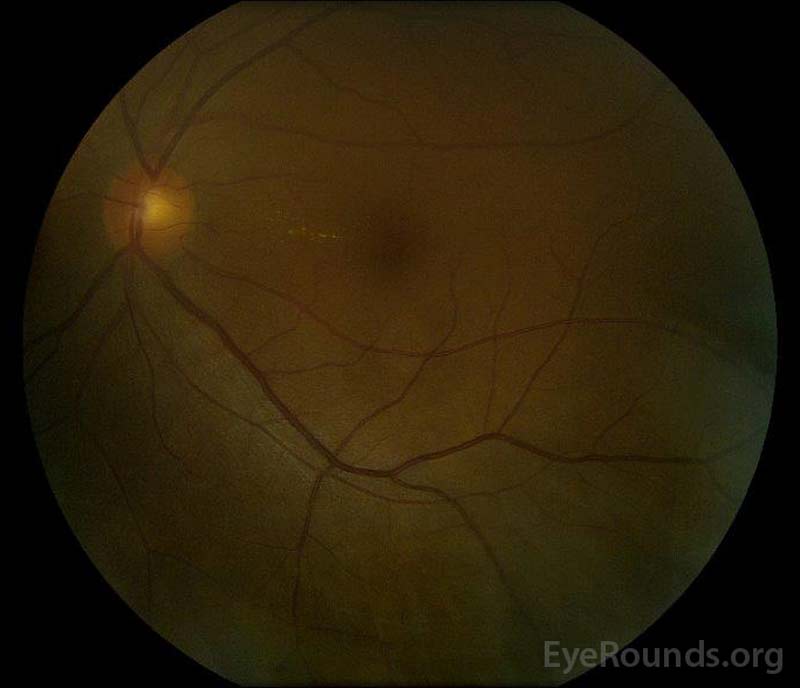 |
Figura 5: Fotografia del fondo a colori degli occhi destro (A) e sinistro (B) durante la fase di convalescenza che dimostra un miglioramento del liquido subretinico e dell’edema del disco.
DIAGNOSI
Incompleta Vogt-Koyanagi-Harada Malattia
DISCUSSIONE
Vogt-Koyanagi-Harada (VKH), la Malattia è una condizione autoimmune sistemica caratterizzata dalla bilaterale, non granulomatosa necrotizzante panuveitis associati con extraoculari tegumentaria modifiche, come poliosis e vitiligine, e l’infiammazione che colpisce l’uvea, interno orecchio, i capelli e meningi. La malattia di Harada è l’uveite isolata senza i segni o sintomi sistemici di VKH.
Eziologia
L’eziologia della malattia di VKH è ancora in gran parte sconosciuta nonostante gli attuali sforzi di ricerca. Si pensa che sia una malattia autoimmune acquisita che coinvolge l’ipersensibilità mediata dalle cellule T agli auto-antigeni melanocitici, con una predisposizione genetica sottostante e un possibile innesco microbico . La tirosinasi ed i peptidi tirosinasi-relativi sono antigeni del melanocita che sono stati suggeriti come obiettivi dei processi autoimmuni in VKH . Tuttavia, l’aumento del rischio di malattia VKH non è stato associato alla famiglia genica della tirosinasi, secondo uno studio .
A causa della maggiore prevalenza tra alcuni gruppi etnici e di genere, si pensa che ci sia una predisposizione genetica nella patogenesi del VKH. Geni multipli, compresi i geni dell’antigene leucocitario umano (HLA) e dell’interleuchina (IL), sono stati associati a VKH in diverse popolazioni etniche . I ricevitori di HLA sono complessi importanti di istocompatibilità in esseri umani che presentano i peptidi al sistema immunitario. HLA-DR1, HLA-DR4, HLA-DRB1 * 0405 e HLA-DRw53 sono diversi aplotipi trovati in pazienti con malattia di VKH . HLA-DR4 è più comune nelle persone giapponesi e ispaniche, mentre HLA-DRB1 * 0405 è più frequente nei pazienti coreani e mediorientali . Sia gli alleli HLA-DR4 che HLA-DRB1*0405 si trovano nei pazienti vietnamiti . Il recettore HLA-DRB1 si lega agli antigeni dei melanociti in diverse capacità. Nonostante queste associazioni, il test genetico non è raccomandato in questo momento.
Dati i soliti sintomi prodromici che accompagnano la VKH, tra cui febbre, mal di testa, meningismo e tinnito, un’eziologia virale incitante è stata suggerita come innesco per l’insorgenza di VKH attraverso meccanismi di mimetismo molecolare in pazienti geneticamente predisposti. La glicoproteina H della busta del citomegalovirus ha omologia significativa dell’aminoacido al peptide della tirosinasi e l’infezione di CMV può innescare VKH con mimetismo molecolare (cioè, riconoscimento dai ricevitori di HLA-classe II). Anche il virus Ebstein-bar (EBV) è stato implicato. Tuttavia, non c’è stata alcuna prova definitiva per quanto riguarda un’eziologia virale di VKH, e non è chiaro che cosa innesca la risposta autoimmune VKH .
Fisiopatologia
Ci sono quattro fasi classiche di VKH che possono avere presentazioni variabili: prodromico, uveitico acuto, convalescente e cronico-ricorrente. I cambiamenti istopatologici iniziano tipicamente nella fase acuta .
La fase uveitica acuta è caratterizzata da ispessimento uveale bilaterale secondario all’infiammazione granulomatosa. I granulomi sono costituiti da linfociti, macrofagi e cellule epitelioidi e giganti piene di granuli . Sebbene le cellule epitelioidi fossero precedentemente ritenute melanociti alterati, uno studio immunoistochimico di follow-up ha suggerito invece un’origine dai macrofagi tissutali . Granulomi pieni di istiociti epitelioidi, chiamati noduli di Dalen-Fuchs, possono spesso essere visti tra l’epitelio pigmentato retinico (RPE) e la membrana di Bruch. L’infiammazione granulomatosa uveale porta ad ispessimento coroidale e distacchi retinici essudativi pieni di liquido proteico. Inoltre, pleocitosi (i.numero di cellule) può essere presente nella camera anteriore e vitreo .
La fase convalescente è identificata dalla depigmentazione delle aree coroide ed extraoculare, compresa la pelle e i capelli perioculari. Una coroide depigmentata impostata contro un nervo ottico pallido dà l’impressione di un fondo “sunset-glow”, che è una caratteristica classica di questa fase di VKH . Inoltre, i noduli di Dalen-Fuchs diventano più prominenti sotto l’RPE nella fase di convalescenza .
La fase cronica-ricorrente è caratterizzata da una diminuzione dello spessore coroidale, risoluzione dei distacchi retinici sierici, vitrite cronica lieve e infiammazione ricorrente del segmento anteriore granulomatoso. La neovascolarizzazione coroidale (CNV) e la fibrosi subretinica possono svilupparsi durante questa fase e sono indicatori di grave progressione della malattia . La cataratta e il glaucoma secondario sono altre complicanze dell’infiammazione di lunga data o ricorrente in questa fase .
Epidemiologia
Il VKH è prevalente nelle razze con pigmento cutaneo più scuro, in particolare asiatici, sudamericani, mediorientali e nativi americani. La malattia di VKH rappresenta > il 10% dell’uveite in queste popolazioni . Solo l ‘ 1-4% dei casi di uveite è considerato secondario alla malattia di VKH negli Stati Uniti (7). Negli Stati Uniti, la maggior parte dei casi di VKH sono stati trovati per colpire individui di asiatici, ispanici e/o nativi americani decenti . È interessante notare che la malattia di VKH colpisce raramente gli africani nonostante la loro pigmentazione scura . L’incidenza della malattia VKH varia notevolmente tra sottogruppi razziali nei paesi vicini . Ad esempio, l’incidenza di VKH in Corea è solo del 2%, molto inferiore a quella riscontrata in Giappone e Cina .
La VKH ha un esordio tipico di età compresa tra 20 e 50 anni ; tuttavia, gli studi suggeriscono che il 3,1-13,4% dei casi di VKH sono pazienti pediatrici e il 10% dei casi ha un’età ≥65 anni . Classicamente, VKH è pensato per avere una predilezione per il genere femminile, e mentre la maggior parte degli studi dimostrano che VKH colpisce in modo sproporzionato le donne, alcuni studi hanno dimostrato una predisposizione maschile o nessuna predisposizione di genere .
Segni / Sintomi
Come già detto, i quattro stadi della malattia di VKH sono prodromici, uveitici, convalescenti e ricorrenti cronici. Ogni fase presenta caratteristiche cliniche distinte.
- Prodromico: Questa fase iniziale può presentarsi come una malattia simil-influenzale con sintomi prevalentemente costituzionali, come mal di testa, vertigini, febbre, affaticamento e/o nausea. Sono stati riportati sintomi neurologici di meningite, paralisi del nervo cranico e neurite ottica, nonché sintomi uditivi di acufene, disacusia e vertigini . Fotofobia, visione offuscata, floater e / o dolore agli occhi di solito iniziano entro 48 ore dai sintomi prodromici . La fase prodromica dura tipicamente da pochi giorni a settimane.
- Uveitico acuto: questa fase include visione sfocata, fotofobia, iniezione congiuntivale e dolore agli occhi. Ci può essere una lieve uveite anteriore che inizialmente appare non granulomatosa. L’insorgenza unilaterale in genere passa al coinvolgimento bilaterale entro 1-2 settimane. Può svilupparsi uveite anteriore granulomatosa con precipitati cheratici di grasso di montone. I risultati dell’esame posteriore possono includere edema del nervo ottico e iperemia, aree multifocali di coroidite, aree multiple di distacchi retinici sierici localizzati al fondo posteriore, ispessimento coroidale, pieghe corioretiniche radianti e vitrite . I distacchi retinici sierosi possono formare un modello di quadrifoglio nel fondo posteriore e potrebbero progredire in ampi distacchi bollosi nei casi più gravi . Il glaucoma infiammatorio acuto è stato associato a questa fase della malattia e può presentarsi con una camera anteriore superficiale secondaria all’edema del corpo ciliare, imitando la chiusura ad angolo acuto . La durata della fase uveitica acuta dipende dalla diagnosi e dalla gestione tempestive.
- Uveitic cronico o convalescente: Questa fase sviluppa tipicamente parecchie settimane dopo la fase acuta ed è caratterizzata dalla vitiligine (per esempio, fronte, mani, spalle, o parte posteriore), dalla poliosi e dall’alopecia . La depigmentazione vicino al limbus corneale, noto come segno di Sugiura, può essere visto un mese dopo l’inizio della malattia ; tuttavia, questo segno è raramente visto al di fuori della popolazione giapponese . La depigmentazione coroidale di solito si verifica nell’arco di pochi mesi e si traduce nel colore rosso-arancio brillante della coroide e nel classico “sunset glow fundus.”Sunset glow fundus è pensato per essere il più importante e predittivo nella diagnosi di VKH cronico . Cicatrici corioretiniche ben definite, rotonde, nummulari possono formarsi nella periferia centrale. La fase uveitica cronica in genere dura diversi mesi.
- Cronico-ricorrente: Questo stadio è caratterizzato da episodi ricorrenti di uveite anteriore granulomatosa con precipitati cheratici di grasso di montone, noduli dell’iride, depigmentazione dell’iride, sinechie posteriori, cataratta subcapsulare posteriore, glaucoma secondario, membrane neovascolari coroidali e, in definitiva, fibrosi subretinica e atrofia corioretinica nummulare . La fase cronica si sviluppa tipicamente almeno sei mesi dopo la presentazione iniziale. I distacchi sierici della retina presenti durante le fasi acute e convalescenti in genere non si ripetono con un trattamento aggressivo con corticosteroidi .
Criteri diagnostici
I criteri diagnostici più recenti, denominati Revised Diagnostic Criteria (RDC) per VKH, sono stati definiti nel 1999 al Primo Workshop internazionale su VKH . Questi sono illustrati nella Tabella 1. I RDC sono utili in quanto dividono VKH in tre categorie diagnostiche differenti basate sulla fase di malattia durante cui un paziente presenta: completo, incompleto e probabile. Questa categorizzazione della malattia consente una gestione appropriata e precoce della malattia ” probabile “che può aiutare a prevenire la progressione della malattia” completa”.
Il work-up per altre cause di infiammazione oculare, sia infettiva che autoinfiammatoria, è essenziale. Questi possono includere la velocità di sedimentazione degli eritrociti (ESR), la proteina C-reattiva (CRP), il test quantiferon-oro per la tubercolosi, la reagina plasmatica rapida (RPR) per la sifilide, l’enzima di conversione dell’angiotensina (ACE) e una radiografia del torace per la sarcoidosi, l’anticorpo antinucleare (ANA) e p-/c-ANCA. Inoltre, una storia di recente trauma oculare o chirurgia intraoculare deve essere notato e probabilmente suggerisce oftalmia simpatica (SO) come la diagnosi più probabile data la presentazione molto simile e fisiopatologia condivisa tra SO e VKH .
Per supportare una diagnosi di VKH in casi equivoci, una puntura lombare può essere eseguita per cercare la pleocitosi linfocitica e monocitica; tuttavia, questo è raramente impiegato clinicamente. L’ottanta per cento dei pazienti ha pleocitosi nel liquido cerebrospinale (CSF) entro una settimana e il 97% ha pleocitosi entro tre settimane. L’aumento dei livelli di cellule immunitarie può durare fino a otto settimane dopo l’insorgenza della malattia . I profili dei marcatori di superficie delle cellule T sono simili tra il CSF e l’umore acqueo, ma diversi dal sangue. Ciò suggerisce la capacità del CSF di riflettere accuratamente l’infiammazione uveale nella malattia di VKH .
Tabella 1. Criteri diagnostici riveduti per la malattia di Vogt-Koyanagi-Harada
*dalla Tabella 1 in (15).
“Malattia di Vogt-Koyanagi-Harada completa (devono essere presenti i criteri da 1 a 5)
- Nessuna storia di trauma oculare penetrante o intervento chirurgico precedente l’insorgenza iniziale di uveite.
- Nessuna evidenza clinica o di laboratorio indicativa di altre entità della malattia oculare.
- Coinvolgimento oculare bilaterale (a o b devono essere soddisfatti, a seconda dello stadio della malattia quando il paziente viene esaminato).
- Manifestazioni precoci della malattia.
- Deve esserci evidenza di una coroidite diffusa (con o senza uveite anteriore, reazione infiammatoria vitrea o iperemia del disco ottico), che può manifestarsi come una delle seguenti:
- Manifestazioni precoci della malattia.
- Aree focali del liquido subretinico o
- Distacchi retinici sierici bollosi.
- Con equivoci fundus risultati; entrambe le operazioni seguenti devono essere presenti, come:
- aree Focali di ritardo nella coroideale perfusione, aree multifocali di individuare perdite, grande placoid aree di hyperfluorescence, di aggregazione all’interno di fluido sottoretinico, nervo ottico e della colorazione (elencati in ordine di comparsa sequenziale) con l’angiografia con fluoresceina e
- Diffusa coroideale ispessimento, senza evidenza di posteriore scleritis con l’ecografia.
- Manifestazioni tardive della malattia.
- Anamnesi indicativa della precedente presenza di reperti da 3a, e sia (2) che (3) sotto o segni multipli da (3):
- Depigmentazione oculare (una delle seguenti manifestazioni è sufficiente): (a) Sunset glow fundus, o (b) Sugiura segno.
- Altri segni oculari:
- Cicatrici depigmentate corioretiniche nummulari, o
- Aggregazione e/o migrazione dell’epitelio pigmentato retinico, o
- Uveite anteriore ricorrente o cronica.
- Risultati neurologici / uditivi (possono essere risolti al momento dell’esame).
- Meningismo (malessere, febbre, mal di testa, nausea, dolore addominale, rigidità del collo e della schiena, o una combinazione di questi fattori; il mal di testa da solo non è sufficiente per soddisfare la definizione di meningismo, tuttavia), o
- Tinnito, o
- Pleiocitosi del liquido cerebrospinale.
- Scoperta tegumentaria (non precedente l’insorgenza della malattia del sistema nervoso centrale o oculare).
- Alopecia, o
- Poliosi, o
- Vitiligine.
Incompleta Vogt-Koyanagi-Harada (malattia di criteri da 1 a 3 e da 4 o 5 devono essere presenti)
- Nessuna storia di penetrazione oculare trauma o un intervento chirurgico precedente l’insorgenza iniziale di uveite, e
- Nessun laboratorio o clinica suggestiva testimonianza di altri oculari enti di malattia, e
- Bilaterali coinvolgimento oculare.
- Reperti neurologici/uditivi; come definito per la malattia di Vogt-Koyanagi-Harada completa sopra, o
- Reperti tegumentari; come definito per la malattia di Vogt-Koyanagi-Harada completa sopra.
Probabile malattia di Vogt-Koyanagi-Harada (malattia oculare isolata; devono essere presenti criteri da 1 a 3)
- Nessuna storia di trauma oculare penetrante o intervento chirurgico precedente l’insorgenza iniziale di uveite.
- Nessuna evidenza clinica o di laboratorio indicativa di altre entità della malattia oculare.
- Coinvolgimento oculare bilaterale come definito per la malattia di Vogt-Koyanagi-Harada completa sopra. “
Test/Work-up di laboratorio
Nel workup iniziale di VKH, si dovrebbe considerare di ottenere i seguenti test:
- Tomografia a coerenza ottica (OCT): Nella fase uveitica acuta, OCT mostrerà probabilmente un significativo ispessimento della coroide e distacchi della retina sierosa. Gli accumuli di liquido subretinico possono avere settazioni che si ritiene siano membrane di fibrina e prodotti infiammatori, creando una struttura lobulare che può essere vista anche sull’angiografia con fluoresceina. Nella fase di convalescenza, OCT può rilevare aree di assottigliamento della retina a seguito di infiammazione risolta dopo il trattamento con corticosteroidi .
- B-ecografia a scansione: Nella fase acuta, l’ecografia può mostrare ispessimento coroidale posteriore diffuso, ispessimento sclerale posteriore, distacchi retinici e opacità vitree . Effusioni ciliari possono essere osservate con biomicroscopia ecografica . Questo test è utile anche per escludere la sclerite posteriore.
- Angiografia della fluoresceina (FA): Classicamente, FA rivela i punti ipofluorescenti coroidali multifocali alla fase iniziale seguita dalle aree iperfluorescenti focali multiple con perdita diffusa nella fase tarda . Il colorante perde attraverso l’RPE e si accumula nello spazio subretinale che circonda i punti iperfluorescenti. FA può essere diagnosticamente utile quando la malattia VKH presenta senza sintomi extraoculari. Iperfluorescenza del disco ottico e difetti della finestra causati da cicatrici corioretiniche atrofiche possono essere visti nella periferia centrale . FA nella fase cronica-ricorrente della malattia VKH mostra difetti della finestra non specifici dovuti a danni RPE, neovascolarizzazione coroidale e fibrosi subretinica .
- Angiografia verde indocianina (ICG) : Fase iniziale ICG raffigura vasi stromali iperfluorescenti che indicano vasculopatia coroidale e punti scuri ipofluorescenti che corrispondono a granulomi e ritardato riempimento a chiazze di vascolarizzazione coroidale . La fase tardiva rivela modelli vascolari stromali fuzzy e iperfluorescenza coroidale diffusa. L’iperfluorescenza del disco è indicativa di una grave malattia. ICGA può rilevare l’infiammazione coroidale subclinica in fasi molto precoci o anche dopo la terapia sistemica .
- Puntura lombare: la pleocitosi nel liquido cerebrospinale è presente nella maggior parte dei pazienti con VKH. La puntura lombare deve essere eseguita all’inizio del decorso della malattia poiché la pleocitosi può risolvere
Trattamento/gestione / Linee guida
Gli obiettivi del trattamento in VKH includono la diagnosi precoce e la soppressione dell’infiammazione attiva, insieme alla prevenzione dell’infiammazione ricorrente e delle complicanze che minacciano la vista, come il glaucoma, il distacco
Il trattamento con corticosteroidi sistemici è la terapia preferita per la malattia di VKH, specialmente durante la fase uveitica acuta. È stato dimostrato che la via di somministrazione di corticosteroidi (orale rispetto a quella endovenosa) non influisce sull’acuità visiva o sull’insorgenza di complicanze visivamente significative nel trattamento del VKH acuto . Per la malattia grave, il protocollo suggerito è la somministrazione endovenosa di metilprednisolone per tre giorni seguita da un trattamento orale con prednisone ad alte dosi. Nella malattia da lieve a moderata, il prednisone orale ad alte dosi può essere sufficiente a 1-2 mg / kg / die. La dose di steroidi deve essere lentamente rastremata per circa sei mesi per prevenire il ripetersi . Il trattamento precoce aggressivo, insieme al test FA seriale che mostra la scomparsa della perdita di colorante attraverso l’RPE, può aiutare a prevenire un’ulteriore progressione della malattia, recidiva e manifestazioni extraoculari . Steroidi topici e cicloplegici possono diminuire le cellule nella camera anteriore e nell’umore vitreo.
Le iniezioni intravitreali e sub-tenoniche di triamcinolone sono state utilizzate per il controllo a breve termine dell’infiammazione intraoculare durante le fasi acute o ricorrenti; queste terapie locali dovrebbero essere considerate nel caso della malattia recalcitrante e nei pazienti che mal tollerano gli effetti collaterali sistemici sfavorevoli degli steroidi dato il cono steroide esteso. Le iniezioni intravitreali anti-VEGF sono talvolta utilizzate per il controllo della neovascolarizzazione coroidale e nei casi di distacchi retinici sierici foveali persistenti .
Gli agenti risparmiatori di steroidi inclusi antimetaboliti, inibitori della calcineurina, farmaci biologici, inibitori del TNF-alfa o agenti citotossici possono essere usati per trattare la VKH e devono essere attentamente monitorati, spesso in coordinamento con un servizio di reumatologia . Vi è stata una discussione in corso sull’uso di agenti immunosoppressori non steroidei come terapia di prima linea per la malattia di VKH. Tuttavia, un recente studio non ha rivelato differenze nei risultati tra il trattamento immunomodulatorio precoce di prima linea (IMT) e il trattamento con prednisone da solo . Inoltre, le terapie immunosoppressive e biologiche sono costose e richiedono un’attenta valutazione pre-trattamento e un frequente follow-up con esami del sangue per valutare gravi effetti collaterali.
Nella fase cronica-ricorrente, la recidiva frequente può suggerire la resistenza alla terapia con corticosteroidi e suggerisce la necessità di un trattamento immunomodulatorio con risparmio di steroidi . L’agente preferito per recidiva resistente agli steroidi o intolleranza agli steroidi è la ciclosporina . Infliximab, rituximab, adalimumab e interferone alfa-2a sono agenti biologici che sono stati anche usati per trattare l’uveite refrattaria nella malattia di VKH.
Per trattare l’uveite anteriore spesso associata a VKH acuto, devono essere prescritti steroidi topici (ad esempio, prednisolone acetato 1%) e cyloplegia topica (ad esempio, ciclopentolato 1% o atropina 1%) a seconda del grado di infiammazione della camera anteriore.
Le complicanze oculari sono comunemente associate alla malattia di VKH. Date le molteplici fasi e la varietà di presentazioni in cui un paziente può presentare con VKH, il trattamento può essere ritardato in molti casi. Nelle forme gravi di VKH e nelle recidive, l’infiammazione intraoculare può essere difficile da controllare e può causare danni strutturali. Oltre il 50% dei pazienti sviluppa complicanze correlate, tra cui cataratta, glaucoma secondario, membrane neovascolari coroidali, fibrosi subretinica o una combinazione di queste (Figura 6) .
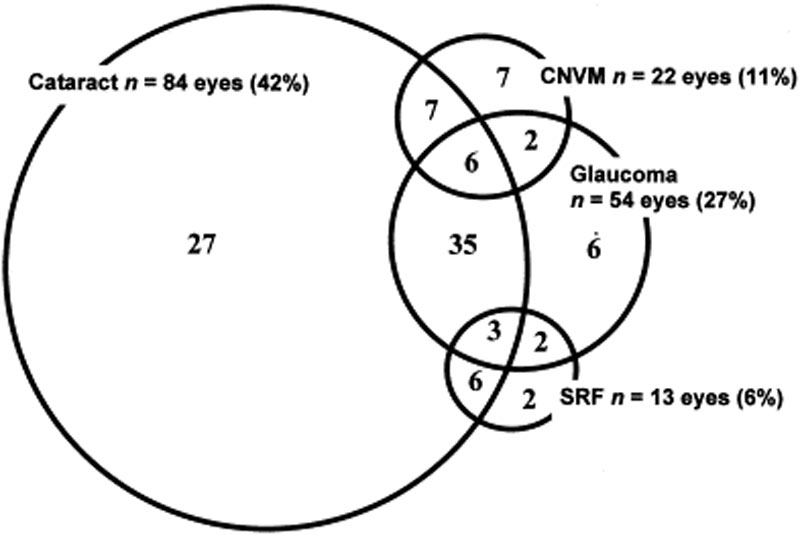
Figura 6: Diagramma di Venn che mostra le complicanze nei pazienti con VKH. (Utilizzato con il permesso di Am J Ophthalmol. 2001;131(5):599-606 )
EPIDEMIOLOGIA ED EZIOLOGIA
|
SIGNS
|
SINTOMI
|
il TRATTAMENTO e la GESTIONE
|
- Du L, Kijlstra A, Yang P. Malattia di Vogt-Koyanagi-Harada: nuove intuizioni in fisiopatologia, diagnosi e trattamento. Prog Retin Res occhio 2016;52:84-111. https://PubMed.gov/26875727. DOI: 10.1016 / j. preteyeres.2016.02.002
- Yamaki K, Gocho K, Hayakawa K, Kondo I, Sakuragi S. Le proteine della famiglia della tirosinasi sono antigeni specifici della malattia di Vogt-Koyanagi-Harada. J Immunol 2000;165 (12): 7323-7329. https://PubMed.gov/11120868
- Horie Y, Takemoto Y, Miyazaki A, Namba K, Kase S, Yoshida K, Has M, Hasumi Y, Inoko H, Mizuki N, Ohno S. Famiglia genica della tirosinasi e malattia di Vogt-Koyanagi-Harada in pazienti giapponesi. Mol Vis 2006;12:1601-1605. https://PubMed.gov/17200659
- Ng JY, Luk FO, Lai TY, Pang CP. Influenza della genetica molecolare nella malattia di Vogt-Koyanagi-Harada. J oftalmica Inflamm Infettare 2014;4: 20. https://PubMed.gov/25097674. DOI: 10.1186 / s12348-014-0020-1
- Bowling B. Uveitis. Kanski clinica oftalmologia New York, New York: Elsevier; 2016; capitolo 11; p. 395-465.
- Yeh PT YC, Yang CH, Lin CP. Distacco retinico nonregmatogeno. In: Schachat AP SS, Hinton DR, Wilkinson CP, Wiedemann P,, editore. La retina di Ryan. New York: Elsevier; 2018; capitolo 99; p. 1828-1849.
- Goto H RK, Rao N. Malattia di Vogt-Koyanagi-Harada. In: Schachat AP SS, Hinton DR, Wilkinson CP, Wiedemann P, editore. La retina di Ryan. New York, New York: Elsevier; 2018; capitolo 78; p. 1505-1515.
- Riddington L, Hall AJ, Tait B, Nicholson I, Varney M. Sindrome di Vogt-Koyanagi-Harada in pazienti di origine vietnamita. Aust N Z J Ophthalmol 1996; 24 (2): 147-149. https://PubMed.gov/9199747
- Sugita S, Takase H, Kawaguchi T, Taguchi C, Mochizuki M. Reazione crociata tra peptidi della tirosinasi e antigene del citomegalovirus da parte delle cellule T di pazienti con malattia di Vogt-Koyanagi-Harada. Int Ophthalmol 2007; 27 (2-3): 87-95. https://PubMed.gov/17253112. DOI: 10.1007 / s10792-006-9020-e
- Freund BK SD, Mieler WF, Yannuzzi LA. Infiammazione. L’Atlante retinico. New York, New York: Elsevier 2017; capitolo 4; p. 279-398.
- Malattia di Rao N. Vogt-Koyanagi-Harada. In: J YMaD, editore. Oculistico. New York, New York: Elsevier; 2014; capitolo 7.17; p. 761-763.
- Rao NA, Xu S, Carattere RL. Oftalmia simpatica. Uno studio immunoistochimico di cellule epitelioidi e giganti. Oftalmologia 1985;92(12):1660-1662. https://PubMed.gov/4088616
- Nussenblatt RB. Sindrome di Vogt-Koyanagi-Harada. In: Whitcup RBNaSM, editore. Uveite: Fondamenti e pratica clinica. 4a Edizione ed: Elsevier; 2010; capitolo Capitolo 24.
- Leggi RW, Holland GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L, Pivetti-Pezzi P, Tessler HH, Usui M. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature. Am J Ophthalmol 2001;131(5):647-652. https://PubMed.gov/11336942
- Chung H, Choi DG. Analisi clinica dell’uveite. Coreano J Ophthalmol 1989; 3(1): 33-37. https://PubMed.gov/2795939. DOI: 10.3341 / kjo.1989.3.1.33
- Abu El-Asrar AM, Al-Kharashi AS, Aldibhi H, Al-Fraykh H, Kangave D. Malattia di Vogt-Koyanagi-Harada nei bambini. Occhio (Lond) 2008;22(9):1124-1131. https://PubMed.gov/17479116. DOI: 10.1038 / sj.occhio.6702859
- Martin TD, Rathinam SR, Cunningham ET. Prevalenza, caratteristiche cliniche e cause di perdita della vista nei bambini con malattia di Vogt-Koyanagi-Harada nell’India meridionale. Retina 2010;30(7):1113-1121. https://PubMed.gov/20168275. DOI: 10.1097 / IAE.0b013e3181c96a87
- Forster DJ, Verde RL, Rao NA. Manifestazione unilaterale della sindrome di Vogt-Koyanagi-Harada in un bambino di 7 anni. Am J Ophthalmol 1991;111(3):380-382. https://PubMed.gov/2000916
- Yamamoto Y, Fukushima A, Nishino K, Koura Y, Komatsu T, Ueno H. Malattia di Vogt-koyanagi-harada con insorgenza in pazienti anziani di età compresa tra 68 e 89 anni. Jpn J Ophthalmol 2007; 51 (1): 60-63. https://PubMed.gov/17295144. DOI: 10.1007 / s10384-006-0379-0
- Wang Y, Chan CC. Differenze di genere nella malattia di vogt-koyanagi-harada e nell’oftalmia simpatica. J Ophthalmol 2014;2014:157803. https://PubMed.gov/24734166. DOI: 10.1155/2014/157803
- Nakao K, Abematsu N, Mizushima Y, Sakamoto T. Gonfiore del disco ottico nella malattia di Vogt-Koyanagi-Harada. Investire Ophthalmol Vis Sci 2012;53 (4): 1917-1922. https://PubMed.gov/22408010. DOI: 10.1167 / iovs.11-8984
- Rao NA, Gupta A, Dustin L, Chee SP, Okada AA, Khairallah M, Bodaghi B, Lehoang P, Accorinti M, Mochizuki M, Prabriputaloong T, Leggi RW. Frequenza delle caratteristiche cliniche distintive nella malattia di Vogt-Koyanagi-Harada. Ophthalmology 2010;117(3):591-599, 599.e591. https://PubMed.gov/20036008. DOI: 10.1016/j.ophtha.2009.08.030
- Veerappan M, Fleischman D, Ulrich JN, Stinnett SS, Jaffe GJ, Allingham RR. The Relationship of Vogt-Koyanagi-Harada Syndrome to Ocular Hypertension and Glaucoma. Ocul Immunol Inflamm 2017;25(6):748-752. https://PubMed.gov/27438521. DOI: 10.1080/09273948.2016.1189578
- Baltmr A, Lightman S, Tomkins-Netzer O. Vogt-Koyanagi-Harada syndrome – current perspectives. Clin Ophthalmol 2016;10:2345-2361. https://PubMed.gov/27932857. DOI: 10.2147/OPTH.S94866
- Kitaichi N, Matoba H, Ohno S. Il ruolo positivo della puntura lombare nella diagnosi della malattia di Vogt-Koyanagi-Harada: sottoinsiemi linfocitari nell’umore acqueo e nel liquido cerebrospinale. Int Ophthalmol 2007; 27 (2-3): 97-103. https://PubMed.gov/17211585. DOI: 10.1007 / s10792-006-9016-7
- Oshima Y, Harino S, Hara Y, Tano Y. Risultati angiografici verdi dell’indocianina nella malattia di Vogt-Koyanagi-Harada. Am J Ophthalmol 1996; 122 (1): 58-66. https://PubMed.gov/8659599
- Leggi RW, Yu F, Accorinti M, Bodaghi B, Chee SP, Fardeau C, Goto H, Holland GN, Kawashima H, Kojima E, Lehoang P, Lemaitre C, Okada AA, Pivetti-Pezzi P, Secchi A, Vedi RF, Tabbara KF, Usui M, Rao NA. Valutazione dell’effetto sui risultati della via di somministrazione di corticosteroidi nella malattia acuta di Vogt-Koyanagi-Harada. Am J Ophthalmol 2006;142(1):119-124. https://PubMed.gov/16815259. DOI: 10.1016 / j. ajo.2006.02.049
- Rubsamen PE, Gass JD. Sindrome di Vogt-Koyanagi-Harada. Decorso clinico, terapia ed esito visivo a lungo termine. Arch Ophthalmol 1991;109(5):682-687. https://PubMed.gov/2025171
- Urzua CA, Velasquez V, Sabat P, Berger O, Ramirez S, Goecke A, Vásquez DH, Gatica H, Guerrero J. Il trattamento immunomodulatorio precedente è associato a migliori risultati visivi in un sottoinsieme di pazienti con malattia di Vogt-Koyanagi-Harada. Acta Ophthalmol 2015;93(6):e475-480. https://PubMed.gov/25565265. DOI: 10.1111 / aos.12648
- Leggi RW, Rechodouni A, Butani N, Johnston R, LaBree LD, Smith RE, Rao NA. Complicanze e fattori prognostici nella malattia di Vogt-Koyanagi-Harada. Am J Ophthalmol 2001;131 (5): 599-606. https://PubMed.gov/11336934
Formato di citazione suggerito
Mai AP, Tran C, Wilson CW, Fox AR, Boldt HC. Malattia di Vogt-Koyanagi-Harada (VKH). EyeRounds.org. Aprile 1, 2019. Disponibile da http://EyeRounds.org/cases/284-vogt-koyanagi-harada.htm
Leave a Reply