Vogt-Koyanagi-Harada (VKH) Sykdom
Forfattere: Anthony P. Mai, BS; Charlene Tran, BS; Caroline W. Wilson, MD; Austin R. Fox, MD; H. Culver Boldt, MD
1.April 2019
FØRSTE PRESENTASJON
Hovedklager
Uklart syn og hodepine
Historie Av Nåværende Sykdom
en 44 ÅR Gammel Vietnamesisk kvinne presentert til beredskapsavdelingen med en 10-dagers Historie Med Progressiv Uklart Syn I BEGGE øyne Og En tre-dagers HISTORIE Med Alvorlig Hodepine. Hennes sentrale synstap hadde ikke forbedret seg med en brytning av hennes optometrist. Hennes alvorlige occipital hodepine forverret med bevegelse og var forbundet med generalisert ubehag, ekstrem tretthet, mild fotofobi og rive. Acetaminophen lindret delvis smerten. Hun hadde nylig reist Til Vietnam, men nektet å møte syke kontakter der. Hun nektet kjeve claudication, feber, eller vektendringer. Hun nektet hudutslett, hørselsendringer, tinnitus, svimmelhet, nummenhet eller prikking. Hun nekter for å ha hatt tuberkulose. Hun hadde ingen historie med tidligere synsproblemer, autoimmune tilstander eller kreft.
Tidligere Okulær Historie
- historie med kosmetisk øyelokkoperasjon (bilateral blepharoplasty) tre år før
- ingen historie med okulært traume eller sykdom
Tidligere Medisinsk Historie
Ingen
Medisiner
Acetaminophen etter behov
Allergier
ingen kjente legemiddelallergier
familiehistorie
ingen historie med øyesykdom eller autoimmun sykdom
sosial historie
hun immigrerte Fra vietnam Flere år før presentasjonen. Hun er gift og har tre barn. Hun jobber på en neglesalong. Hun bruker ikke tobakksprodukter, alkohol eller ulovlige stoffer. Hun reiser til Vietnam hver sjette til tolv måneder.
Gjennomgang Av Systemer
Negativ bortsett fra det som er detaljert i historien om nåværende sykdom
OKULÆR UNDERSØKELSE
Synsskarphet med/uten korreksjon (Snellen)
- Høyre øye (OD): 20/300 (ingen forbedring med pinhole)
- Venstre øye (OS): 20/60-2+2 (ingen forbedring med pinhole)
Okulær Motilitet/Justering
fulle ekstraokulære bevegelser i begge øyne (OU)
Intraokulært Trykk (IOP): (Tonopen)
- OD: 12 mmHg
- OS: 14 mmHg
Elever
- OD: 4 mm i mørke, 3 mm i lys, ingen relativ afferent pupilldefekt (RAPD)
- OS: 4 mm i mørke, 3 mm i lys, ingen RAPD
Konfrontasjon visuelle felt: (Count OD: CENTRAL SCOTOMA
ekstern
normal på begge sider
slit lampe eksamen
- lokk/VIPPER: normal ou
- conjunctiva/sclera: klar OG rolig ou
- hornhinnen: 1+ punktlig epiteliale erosjoner, ingen keratiske utfellinger OU
- fremre kammer: Spor celle og fakkel og dyp Ou
- Iris: Normal arkitektur OU
- Linse: Klar OU
Dilatert fundus undersøkelse (DFE)
- Glasslegemet: Spor fremre glasslegemet celler OU
- Plate:
- OD: Grad 3 diskødem, hyperemisk
- os: grad 2-3 diskødem, hyperemisk
- kopp-til-disk-forhold: 0,0 OU
- makula:
- od: 3 + Cystoid makulært ødem (CME) og subretinal væske (srf) som strekker seg fra Platen Til den temporale makula. Ingen lipid eller ekssudater. Boggy-vises choroid.
- OS: 2 + CME og SRF som strekker seg fra platen gjennom fovea. 1-2 + lineær lipid som strekker seg fra platen mot fovea. Boggy-vises choroid.
- Fartøy:
- Od: Kappe temporært
- Os: Normal
- Periferi:
- OD: Cystisk retinal tuft anterior til ekvator ved 10:30
- OS: Grunne SRF anterior til ekvator ved 4:00
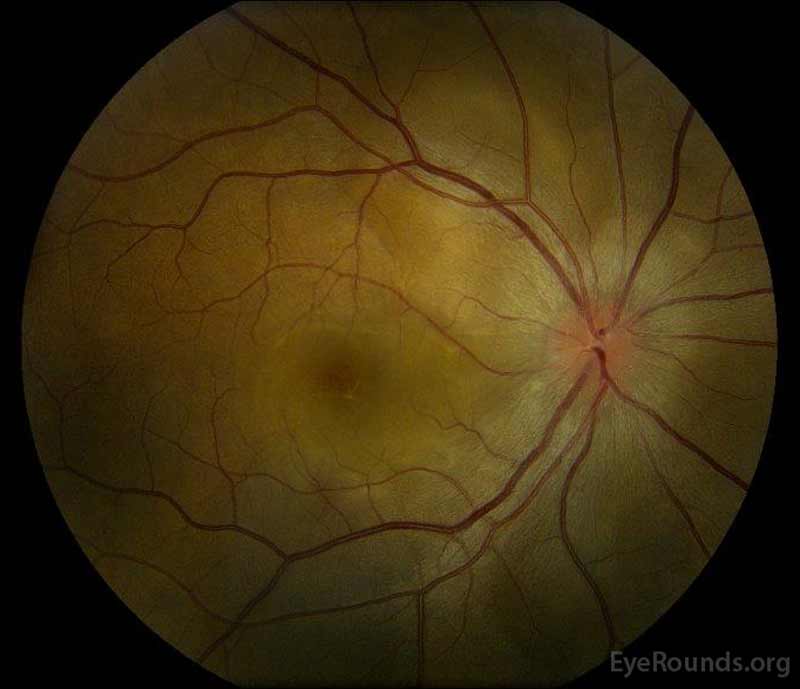
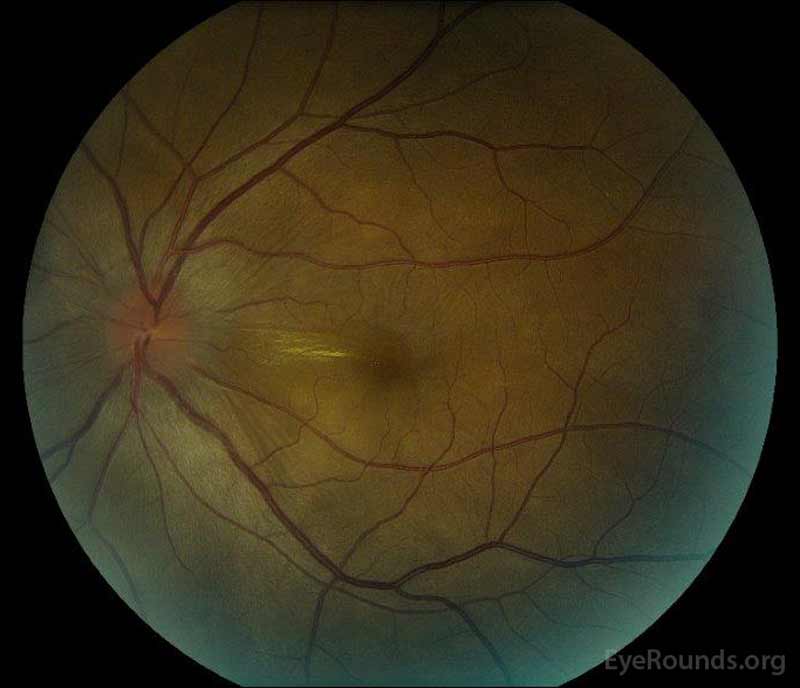
figur 1: farge fundus fotografier ved presentasjon: (venstre bilde) høyre øye har skiveødem og mild hyperemi samt subretinal væske som strekker seg fra platen midlertidig gjennom makulaen. Det er også en fokal serøs retinal detachment superotemporal til platen, langs overlegen arkade. (Right image) The left eye has disc edema and mild hyperemia, along with subretinal fluid extending from the disc to the macula and linear lipid deposits in the nasal macula.
 |
 |
 |
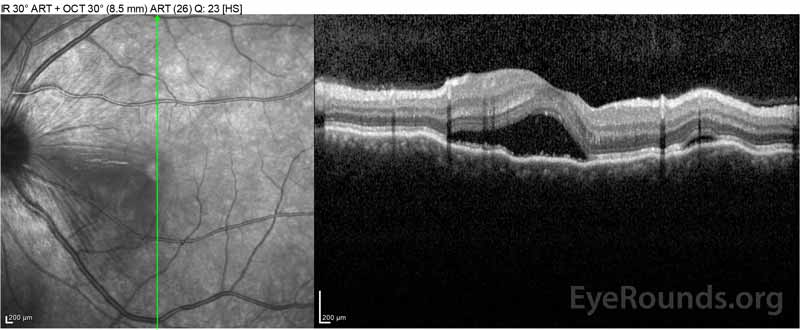 |
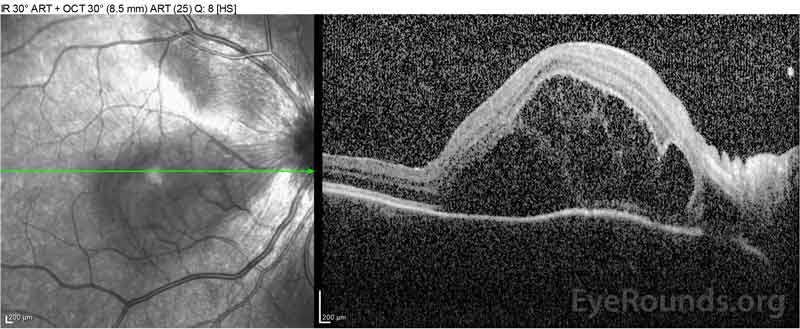
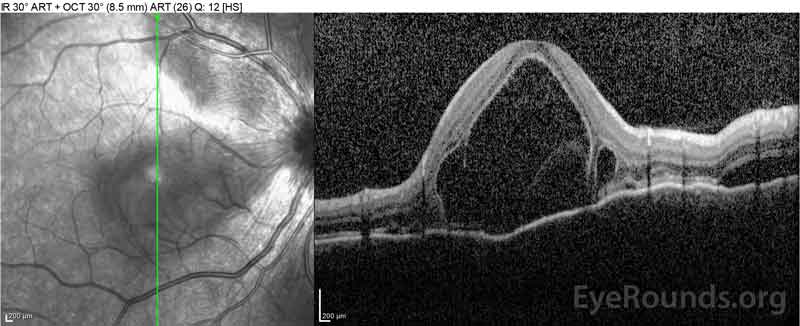
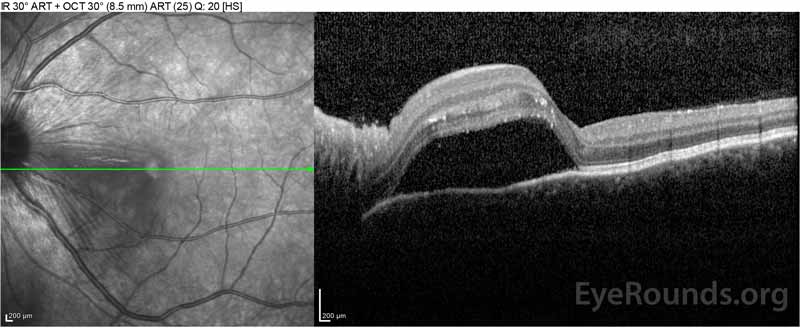
Figure 2: Optisk koherens tomografi (OCT) i høyre øye (øvre paneler) viser en serøs retinal detachment som involverer fovea med omfattende overliggende intraretinal væske, forstyrrelse av de ytre retinale lagene og undulations av den fortykkede choroid. OCT i venstre øye (bunnpaneler) viser en serøs retinal detachment i nasal makula som strekker seg opp til fovea.
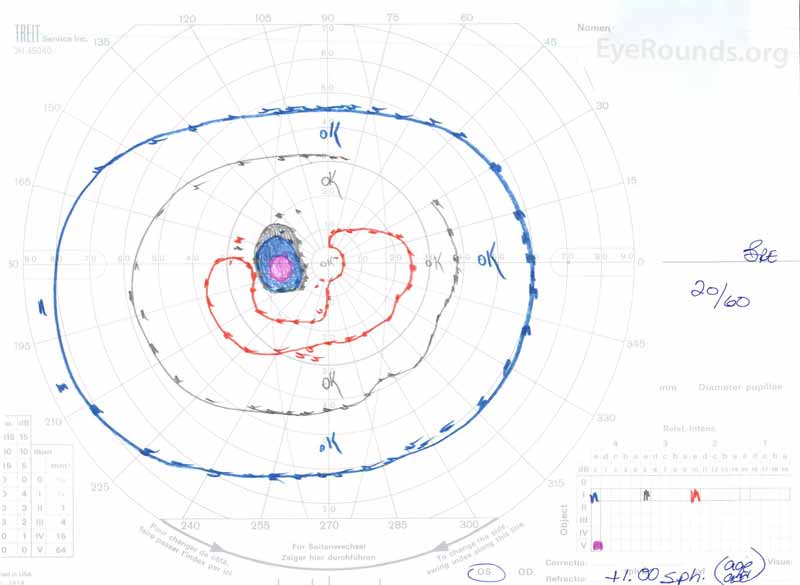
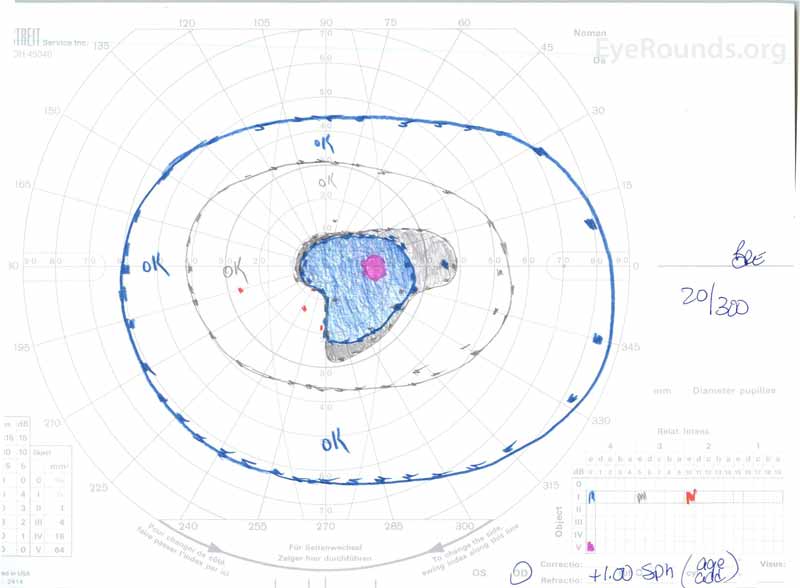
figur 3: Goldman visuelle felt (GVF), OU. (Venstre bilde) OS viser en forstørret fysiologisk blind flekk og mild sentral scotoma. (Høyre bilde) od viser en moderat tett sentral scotoma innlemme fysiologisk blind flekk og utvide inferotemporally.
B-scan: Ingen tegn på skleritt, milde vitreale opasiteter/celler inferior
Differensialdiagnose
- Akutt posterior multifokal placoid pigmentepiteliopati (APMPPE)
- Sentral serøs chorioretinopati
- Optisk neuritt
- Panuveitt
- Autoimmun sykdom (F.eks. Sle, sarkoidose)
- infeksjon (E.G, syfilis, tuberkulose, Bartonella henselae)
- Malignitet (f. eks. okulært lymfom)
- Posterior skleritt
- Sympatisk oftalmia
- Uveal effusjonssyndrom
- Vogt-Koyanagi-Harada Syndrom
OPPARBEIDING
komplett blodtelling
antall hvite blodlegemer: 4,9 k/mm3 (ref: 3,7-10,5)
antall røde blodlegemer 3,99 m/mm3 (ref: 4,0-5,2)
hemoglobin 11,6 g/Dl (Ref: 11,9-15,5)
hematokrit 35 % (ref: 35-47)
Basic metabolic panel
Sodium 138 mEq/L (Ref: 135-145)
Potassium 4.3 mEq/L (Ref: 3.5-5.0)
Chloride 107 mEq/L (Ref: 95-107)
CO2 20 mEq/L (Ref: 22-29)
Blood urea nitrogen 16 mEq/dL (Ref: 10-20)
Creatinine 0.7 mg/dL (Ref: 0.5-1.0)
C-reactive Protein (CRP): <0.5 mg/dL (Ref: <=0.5)
Erythrocyte sedimentation rate (ESR): 12 mm/Hr (Ref: 0-20)
Angiotensin–converting enzyme (ACE): 13 U/L (Ref: 8-52)
QuantiFERON-TB Gull: negativ
Jern, blod 54 mikrogram / dL (Ref: 37-145)
Total jernbindingskapasitet 379 mikrogram/dL (Ref: 250-425)
KLINISK KURS
pasienten ble først evaluert av akuttmottaket gitt hennes klager av ny-onset alvorlig hodepine og synstap. Hjernen computertomografi (CT) og magnetisk resonans imaging (MRI) skanner var unremarkable. ESR og CRP var innenfor normale nivåer. Oftalmologiklinikken evaluerte henne dagen etter og fant bilaterale serøse retinale løsninger og panuveitt. ACE Og QuantiFERON-TB Gold labs var begge negative. Hun ble diagnostisert Med Vogt-Koyanagi-Harada sykdom basert på hennes kliniske presentasjon og Asiatisk avstamning. Hun ble behandlet med 80 mg prednison daglig, acetaminophen etter behov for hodepine, Og Vitamin D og kalsiumtilskudd. Hennes hodepine raskt løst, og hennes synsskarphet stadig bedre i løpet av de neste to ukene. Hennes prednison dosering ble deretter avsmalnet ned til 40 mg over tre uker med fortsatt oppløsning av symptomer og forbedring i synsskarphet. Hun hadde ingen tilbakefall av hodepine eller forverret syn under prednison taper. På hennes siste avtale, hun hadde avtatt ned til 5 mg annenhver dag, uten retur av symptomer. Hennes synsskarphet ved det oppfølgingsbesøket var 20/15-2 OD og 20/20 + 2 OS, og macular OCT viste full oppløsning av skiveødem og serøs retinal detachments i begge øynene (Figur 4).
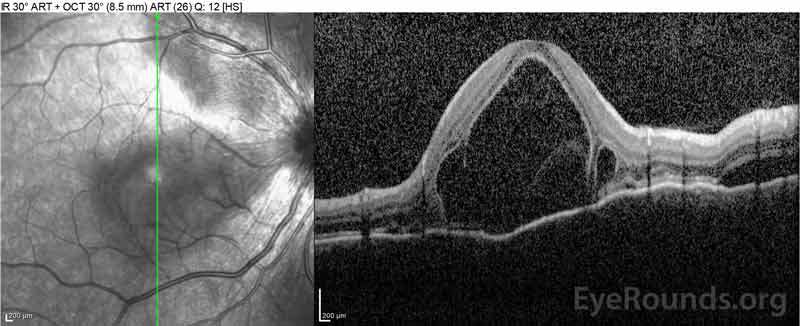
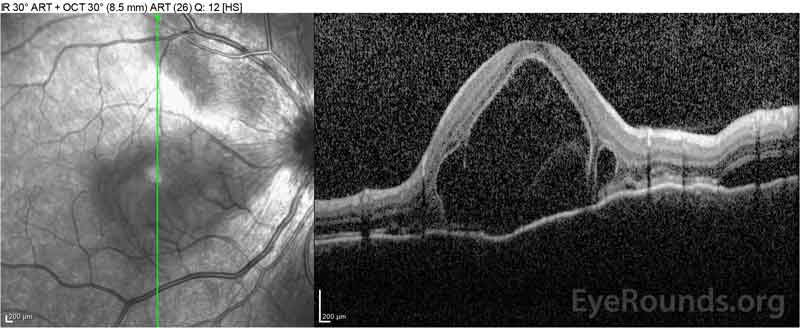
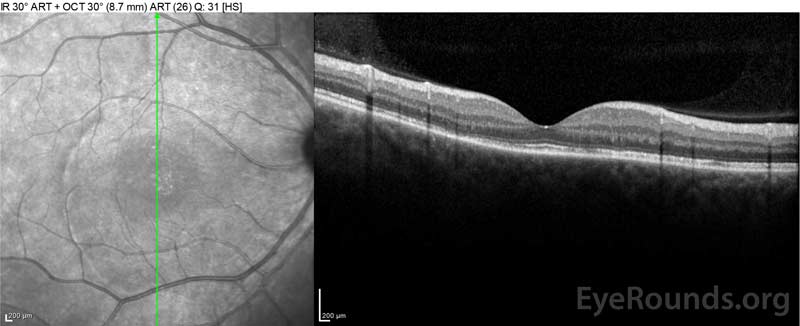
Figur 4: Optisk koherens-tomografi som viser subretinalvæske ved baseline (øverst) og oppløsningsforløpet ved en uke (mellom) og fem uker (nederst) mens du er på en høydose oral prednison taper. Legg merke til utjevning av koroidale undulasjoner med behandling.
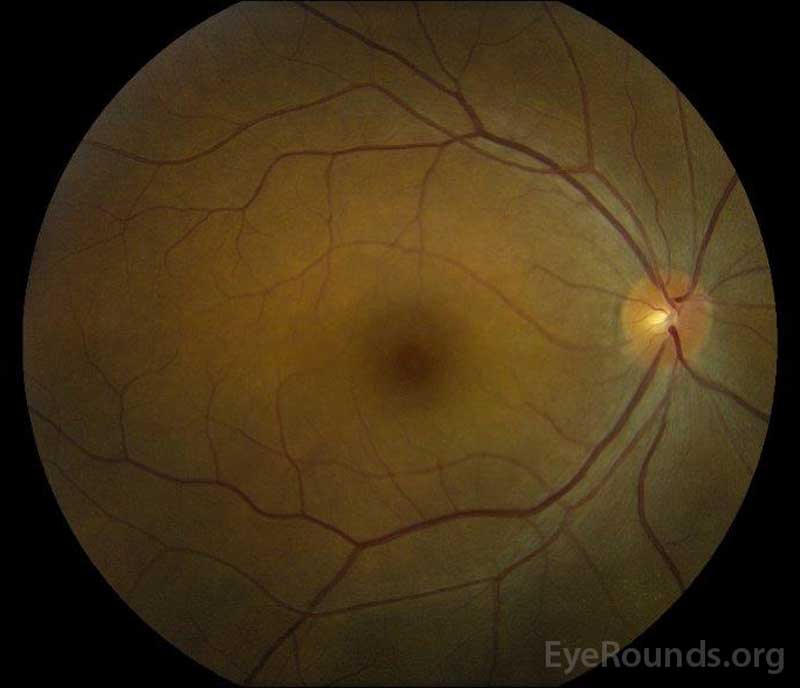 |
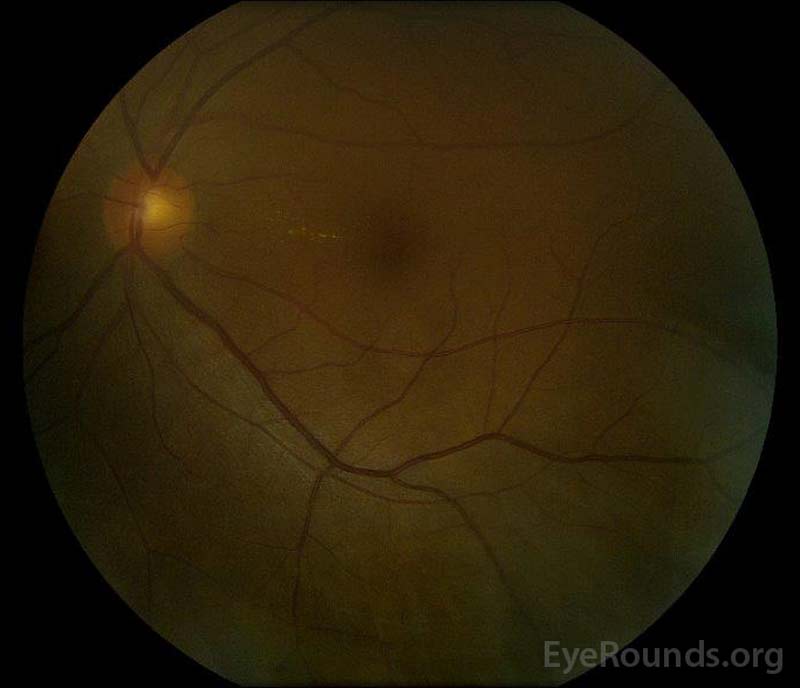
figur 5: Color fundus fotografering av høyre (A) og venstre (b) øyne under rekonvalesens fase demonstrere forbedring i subretinal væske og plate ødem.
DIAGNOSE
Ufullstendig Vogt-Koyanagi-Harada Sykdom
DISKUSJON
Vogt-Koyanagi-HARADA (VKH) Sykdom er en systemisk autoimmun tilstand preget av bilateral ikke-nekrotiserende granulomatøs panuveitt forbundet med ekstraokulære integumentære endringer, som poliose og vitiligo, og betennelse som påvirker uvea, indre øre, hår og meninges. Harada sykdom er isolert uveitt uten systemiske tegn eller symptomer PÅ VKH.
Etiologi
etiologien TIL VKH sykdom er fortsatt stort sett ukjent til tross for dagens forskningsinnsats. Det antas å være en ervervet autoimmun sykdom som involverer t-celle-mediert overfølsomhet overfor melanocytiske selvantigener, med en underliggende genetisk predisponering og mulig mikrobiell utløser . Tyrosinase og tyrosinase-relaterte peptider er melanocyttantigener som har blitt foreslått som mål for autoimmune prosesser I VKH . Økt risiko FOR VKH-sykdom var imidlertid ikke forbundet med tyrosinasegenfamilien, ifølge en studie .
på grunn av økt forekomst blant visse etniske og kjønnsgrupper, antas det å være en genetisk predisposisjon i patogenesen av VKH. Flere gener, inkludert humant leukocyttantigen (hla) og interleukin (IL) gener, har vært assosiert MED VKH i ulike etniske populasjoner . Hla-reseptorer er store histokompatibilitetskomplekser hos mennesker som presenterer peptider til immunsystemet. HLA-DR1, HLA-DR4, HLA-DRB1 * 0405 og HLA-DRw53 er flere haplotyper funnet hos pasienter med VKH sykdom . HLA-DR4 er mer vanlig Hos Japanske og Spanske mennesker, MENS HLA-DRB1 * 0405 er hyppigere hos koreanske og Midtøsten pasienter . Både hla-DR4 og HLA-DRB1*0405 alleler finnes Hos Vietnamesiske pasienter . Hla-DRB1-reseptoren binder seg til melanocyttantigener i varierende kapasiteter. Til tross for disse foreningene anbefales ikke genetisk testing på dette tidspunktet. Gitt de vanlige prodromale symptomene som følger MED VKH, inkludert feber, hodepine, meningismus og tinnitus, har en fremkallende viral etiologi blitt foreslått som en utløser for VKH-utbrudd gjennom mekanismer for molekylær etterligning hos genetisk predisponerte pasienter. Cytomegalovirus konvoluttglykoprotein H har signifikant aminosyrehomologi til tyrosinasepeptidet, OG CMV-infeksjon kan utløse VKH gjennom molekylær etterligning (dvs .gjenkjenning av hla-klasse II-reseptorer). Ebstein-bar virus (EBV) har også vært involvert. Det har imidlertid ikke vært noe endelig bevis på en viral etiologi AV VKH, og det er fortsatt uklart hva som utløser vkh autoimmun respons .
Patofysiologi
det er fire klassiske faser AV VKH som kan ha variable presentasjoner: prodromal, akutt uveittisk, konvalescerende og kronisk tilbakevendende. Histopatologiske endringer begynner vanligvis i den akutte fasen .
den akutte uveittiske fasen er preget av bilateral uveal fortykkelse sekundært til granulomatøs betennelse. Granulomene består av lymfocytter, makrofager og granulatfylte epitelioide og gigantiske celler . Selv om epitelioidcellene tidligere ble antatt å være endret melanocytter, foreslo en oppfølgings immunhistokjemisk studie en opprinnelse fra vevsmakrofager i stedet . Granulomer fylt med epitelioide histiocytter, Kalt Dalen-Fuchs noduler, kan ofte ses mellom retinalpigmentepitelet (RPE) og Bruchs membran. Den uveale granulomatøse betennelsen fører til koroidal fortykkelse og eksudativ retinal detachments fylt med proteinholdig væske. I tillegg pleocytose (i.e., økt celletall) kan være til stede i det fremre kammer og glasslegemet .
rekonvalesentfasen er identifisert ved depigmentering av choroid og ekstraokulære områder, inkludert periokulær hud og hår. En depigmentert choroid sett mot en blek optisk nerve gir inntrykk av en» sunset-glow » fundus, som er et klassisk trekk ved DENNE fasen AV VKH . I Tillegg blir Dalen-Fuchs knuter mer fremtredende under RPE i rekonvalesensfasen .
kronisk tilbakevendende fase er karakterisert ved redusert koroidal tykkelse, oppløsning av serøs retinal detachments, kronisk mild vitritis, og tilbakevendende granulomatøs anterior segment betennelse. Koroidal neovaskularisering (CNV) og subretinal fibrose kan utvikles i denne fasen og er indikatorer på alvorlig sykdomsprogresjon . Katarakt og sekundær glaukom er andre komplikasjoner av langvarig eller tilbakevendende betennelse i denne fasen .
Epidemiologi
VKH er utbredt i løp med mørkere hudpigment, spesielt Asiater, Søramerikanere, Midtøsten Og Indianere. VKH sykdom står for > 10% av uveitt i disse populasjonene . Bare 1-4% av uveitt tilfeller antas å være sekundær TIL VKH sykdom i Usa (7). I Usa har de fleste tilfeller AV VKH blitt funnet å påvirke personer Av Asiatisk, Spansk Og / Eller Indiansk anstendig . Interessant, PÅVIRKER VKH sykdom Sjelden Afrikanere til tross for deres mørke pigmentering . Forekomsten av VKH sykdom varierer sterkt mellom rasemessige undergrupper i nabolandene . For Eksempel Er Koreas forekomst AV VKH bare 2%, mye lavere enn det som finnes I Japan og Kina .
VKH har et typisk utbrudd på 20 til 50 år , men studier tyder på at 3,1-13,4% av vkh-tilfellene er pediatriske pasienter og 10% av tilfellene er ≥65 år . Klassisk ANTAS VKH å ha en forkjærlighet for det kvinnelige kjønn, og mens de fleste studier viser at VKH uforholdsmessig rammer kvinner, har noen studier vist en mannlig predisposisjon eller ingen kjønnsperspektiv .
Tegn / Symptomer
som nevnt ovenfor er DE fire stadiene AV VKH sykdom prodromal, uveittisk, rekonvalesent og kronisk tilbakevendende. Hvert stadium viser forskjellige kliniske egenskaper.
- Prodromal: denne innledende fasen kan presentere som en influensalignende sykdom med overveiende konstitusjonelle symptomer, som hodepine, svimmelhet, feber, tretthet og/eller kvalme. Nevrologiske symptomer på hjernehinnebetennelse, hjernenervepalsies, og optisk nevritt, samt auditive symptomer på tinnitus, dysacusis, og vertigo er rapportert . Fotofobi, sløret syn, flytere og/eller øyesmerter begynner vanligvis innen 48 timer etter prodromale symptomer . Prodromalfasen varer vanligvis fra noen dager til uker.
- Akutt Uveittisk: Dette stadiet inkluderer uklart syn, fotofobi, konjunktivinjeksjon og øyesmerter. Det kan være mild fremre uveitt som først vises ikke-granulomatøs. Unilateral debut går vanligvis over til bilateral involvering innen 1-2 uker. Granulomatøs anterior uveitt med fårekjøtt-fett keratiske utfellinger kan utvikle seg. Bakre undersøkelsesfunn kan omfatte optisk nerveødem og hyperemi, multifokale områder av choroiditt, flere områder av serøs retinal detachments lokalisert til bakre fundus, choroidal fortykkelse, utstrålende chorioretinale folder og vitritis . Serøs netthinneavløsning kan danne et kløverblad mønster i bakre fundus og kan utvikle seg til omfattende bulløse løsninger i alvorlige tilfeller . Akutt inflammatorisk glaukom har vært assosiert med denne fasen av sykdommen og kan presentere med et grunt fremre kammer sekundært til ciliary kropps ødem, etterligne akutt vinkel lukning . Varigheten av den akutte uveittiske fasen avhenger av rask diagnose og ledelse. Kronisk Uveittisk Eller Rekonvalesent: dette stadiet utvikler seg vanligvis flere uker etter den akutte fasen og er preget av vitiligo (f .eks. ansikt, hender, skuldre eller rygg), poliose og alopecia. Depigmentering nær hornhinnen limbus, kjent som Sugiura ‘ s sign, kan ses en måned etter sykdomsutbrudd ; dette tegnet er imidlertid sjelden sett utenfor Den Japanske befolkningen . Choroidal depigmentering oppstår vanligvis over noen måneder og resulterer i den lyse oransje-røde fargen på choroid og den klassiske «sunset glow fundus.»Sunset glow fundus antas å være den viktigste og prediktive i diagnosen kronisk VKH . Veldefinerte, runde, nummulære chorioretinale arr kan danne seg i midten av periferien. Den kroniske uveittiske fasen varer vanligvis flere måneder.
- Kronisk tilbakevendende: Dette stadiet er preget av tilbakevendende episoder av granulomatøs fremre uveitt med fårekjøttfett keratisk utfelling, iris noduler, iris depigmentering, posterior synechiae, posterior subkapsulær katarakt, sekundær glaukom, choroidal neovaskulær membraner, og til slutt subretinal fibrose og nummular chorioretinal atrofi . Den kroniske fasen utvikler seg vanligvis minst seks måneder etter første presentasjon. De serøse netthinneavløsningene som er tilstede under akutte og rekonvalesente faser, oppstår vanligvis ikke igjen ved aggressiv kortikosteroidbehandling .
Diagnostiske Kriterier
De siste diagnostiske kriteriene, Kalt De Reviderte Diagnostiske Kriteriene (RDC) FOR VKH, ble definert i 1999 på Den Første Internasjonale Workshopen OM VKH . Disse er beskrevet I Tabell 1. RDC er nyttig ved at de deler VKH i tre forskjellige diagnostiske kategorier basert på sykdomsfasen der en pasient presenterer: komplett, ufullstendig og sannsynlig. Denne kategoriseringen av sykdom muliggjør hensiktsmessig og tidlig styring i «sannsynlig» sykdom som kan bidra til å forhindre progresjon til» komplett » sykdom.
Arbeid opp for andre årsaker til okulær betennelse, både smittsomme og auto-inflammatorisk, er avgjørende. Disse kan omfatte senkning (ESR), C-reaktivt protein (CRP), quantiferon-Gull testing for tuberkulose, rapid plasma reagin (RPR) for syfilis, angiotensin-converting enzyme (ACE) og en brystet x-ray for sarkoidose, antinukleære antistoff (ANA), og p-/c-ANCA. Også en historie med nylig okulært traume eller intraokulær kirurgi må noteres og sannsynligvis antyder sympatisk oftalmia (SO) som den mer sannsynlige diagnosen gitt den svært liknende presentasjonen og patofysiologien som deles MELLOM SO og VKH .
for å støtte EN DIAGNOSE AV VKH i tvetydige tilfeller kan en lumbal punktering utføres for å se etter lymfocytisk og monocytisk pleocytose; dette er imidlertid sjelden ansatt klinisk. Åtti prosent av pasientene har pleocytose i cerebrospinalvæsken (CSF) innen en uke og 97% har pleocytose innen tre uker. Økte nivåer av immunceller kan vare opptil åtte uker etter sykdomsutbrudd . T-cellens overflatemarkørprofiler er like mellom CSF og vandig humor, men forskjellig fra blodet. DETTE antyder CSFS evne til nøyaktig å reflektere uveal betennelse I VKH sykdom .
Tabell 1. Reviderte Diagnostiske Kriterier For Vogt-Koyanagi-Harada Sykdom
* Fra Tabell 1 i (15).
«Komplett Vogt-Koyanagi-Harada sykdom (kriterium 1 til 5 må være tilstede)
- ingen anamnese med penetrerende okulært traume eller kirurgi før den første utbruddet av uveitt.
- Ingen kliniske eller laboratoriebevis som tyder på andre okulære sykdommer.
- Bilateral okulær involvering(a eller b må oppfylles, avhengig av sykdomsstadiet når pasienten undersøkes).
- Tidlige manifestasjoner av sykdom.
- det må være tegn på diffus choroiditt (med eller uten fremre uveitt, vitreøs inflammatorisk reaksjon eller optisk diskhyperemi), som kan manifestere seg som ett av følgende:
- Fokale Områder av subretinal væske, eller
- bullous serøs retinal detachments.
- med tvetydige fundus funn; begge de følgende må være til stede i tillegg:
- Fokale områder av forsinkelse i koroidal perfusjon, multifokale områder av pinpoint lekkasje, store placoid områder av hyperfluorescens, pooling innenfor subretinal væske, og synsnervefarging (oppført i rekkefølge av sekvensiell utseende) ved fluorescein angiografi, og
- Diffus koroidal fortykkelse, uten tegn på posterior skleritt ved ultralyd.
- Sene manifestasjoner av sykdom.
- Historie som tyder på tidligere funn fra 3a, og enten både (2) og (3) under eller flere tegn fra (3):
- okulær depigmentering (en av følgende manifestasjoner er tilstrekkelig): (A) Sunset glow fundus, eller (b) Sugiura tegn.
- Andre okulære tegn:
- Nummular chorioretinal depigmented arr, eller
- netthinnepigmentepitel klumping og/eller migrasjon, eller
- Tilbakevendende eller kronisk fremre uveitt.
- Nevrologiske / auditive funn (kan ha løst ved undersøkelsestidspunktet).
- Meningismus (sykdomsfølelse, feber, hodepine, kvalme, magesmerter, stivhet i nakke og rygg, eller en kombinasjon av disse faktorene; hodepine alene er ikke tilstrekkelig til å møte definisjonen av meningismus, derimot), Eller
- Tinnitus, Eller
- Cerebrospinalvæske pleocytose.
- integumentary funn (ikke før utbruddet av sentralnervesystemet eller okulær sykdom). Det kan være Vanskelig å finne En Løsning på Problemet.
- ingen anamnese med penetrerende okulært traume eller kirurgi før den første utbruddet av uveitt, Og
- Ingen kliniske eller laboratoriemessige bevis som tyder på andre okulære sykdommer, Og
- Bilateral okulær involvering.
- Nevrologiske / auditive funn; som definert for Komplett Vogt-Koyanagi-Harada sykdom ovenfor, eller
- Integumentære funn; som definert for komplett Vogt-Koyanagi-Harada sykdom ovenfor.
- ingen anamnese med penetrerende okulært traume eller kirurgi forut for den første utbruddet av uveitt.
- Ingen kliniske eller laboratoriebevis som tyder på andre okulære sykdommer.
- Bilateral okulær involvering som definert for fullstendig Vogt-Koyanagi-Harada sykdom ovenfor. «
Ufullstendig Vogt-Koyanagi-harada-sykdom (kriterium 1 til 3 og enten 4 eller 5 må være tilstede)
Sannsynlig Vogt-Koyanagi-Harada sykdom (isolert okulær sykdom; kriterium 1 til 3 må være tilstede)
Testing / Laboratoriearbeid
I DEN første opparbeidelsen AV VKH bør man vurdere å oppnå følgende tester:
- Optisk koherens tomografi (OCT): I den akutte uveittfasen vil OCT trolig vise signifikant koroidal fortykkelse og serøs retinal detachments. De subretinale væskeakkumulasjonene kan ha septasjoner som antas å være fibrinmembraner og inflammatoriske produkter, noe som skaper en lobulær struktur som også kan ses på fluoresceinangiografi. I rekonvalesensfasen kan OCT påvise områder av retinal tynning etter løst betennelse etter kortikosteroidbehandling .
- B-skanning ultralyd: I den akutte fasen kan ultralyd vise diffus posterior koroidal fortykkelse, posterior skleral fortykkelse, retinale løsrivelser og glassformede opasiteter . Ciliær effusjon kan observeres ved ultralydbiomikroskopi . Denne testen er også nyttig for å utelukke bakre skleritt. Fluoresceinangiografi (FA): Klassisk avslører FA multifokale choroidale hypofluorescerende prikker i tidlig fase etterfulgt av flere fokale hyperfluorescerende områder med diffus lekkasje i sen fase . Fargestoffet lekker gjennom RPE og akkumuleres i det subretinale rommet som omgir de hyperfluorescerende prikkene. FA KAN være diagnostisk nyttig når VKH sykdom presenterer uten ekstraokulære symptomer. Optisk plate hyperfluorescens og vindu defekter forårsaket av atrofisk chorioretinal arr kan sees i midten av periferien . FA i kronisk tilbakevendende STADIUM AV VKH sykdom viser uspesifikke vindusfeil på GRUNN AV rpe-skade, koroidal neovaskularisering og subretinal fibrose .
- Indocyanin grønn (ICG) angiografi: TIDLIG FASE ICG viser hyperfluorescent stromal fartøy som indikerer choroidal vaskulopati og hypofluorescent mørke prikker som tilsvarer granulomer og forsinket usammenhengende fylling av choroidal vaskulatur . Den sene fasen avslører fuzzy stromale vaskulære mønstre og diffus choroidal hyperfluorescens. Disc hyperfluorescens tyder på alvorlig sykdom. ICGA kan oppdage subklinisk koroidal betennelse i svært tidlige stadier eller til og med etter systemisk terapi .
- Lumbar punktering: Pleocytose i cerebrospinalvæsken er tilstede hos de fleste VKH-pasienter. Lumbalpunksjon bør utføres tidlig i sykdomsforløpet siden pleocytose kan løse
Behandling/Ledelse / Retningslinjer
behandlingsmål i VKH inkluderer tidlig diagnose og undertrykkelse av aktiv betennelse, sammen med forebygging av tilbakevendende betennelse og synstruende komplikasjoner, som glaukom, bulløs retinal detachment og koroidal neovaskularisering.
Systemisk kortikosteroidbehandling er den foretrukne behandlingen for VKH-sykdom, spesielt i det akutte uveittiske stadiet. Det er vist at administrering av kortikosteroider (oral versus intravenøs) ikke påvirker synsskarpheten eller forekomsten av visuelt signifikante komplikasjoner ved behandling av akutt VKH . Ved alvorlig sykdom er den foreslåtte protokollen intravenøs administrering av metylprednisolon i tre dager etterfulgt av oral høydose prednison-behandling. Ved mild moderat sykdom kan høydose oral prednison være tilstrekkelig ved 1-2 mg/kg / dag. Steroiddosen bør sakte reduseres over omtrent seks måneder for å forhindre tilbakefall . Aggressiv tidlig behandling, sammen med seriell FA-testing som viser forsvinning av fargestofflekkasje gjennom RPE, kan bidra til å forhindre ytterligere sykdomsprogresjon, tilbakefall og ekstraokulære manifestasjoner . Aktuelle steroider og cycloplegics kan redusere celler i fremre kammer og glasslegem. Intravitreale og sub-Tenon injeksjoner av triamcinolon har blitt brukt for kortvarig kontroll av intraokulær betennelse i akutte eller tilbakevendende faser; disse lokale terapiene bør vurderes i tilfelle av tilbakevendende sykdom og hos pasienter som dårlig tolererer de ugunstige systemiske bivirkningene av steroider gitt den utvidede steroidtapet. Intravitreale anti-VEGF injeksjoner brukes noen ganger for kontroll av koroidal neovaskularisering og i tilfeller av vedvarende foveal serøs retinal detachments .
Steroidsparende midler, inkludert antimetabolitter, kalsinevrinhemmere, biologiske legemidler, tnf-alfa-hemmere eller cytotoksiske midler, kan brukes til å behandle VKH og bør overvåkes nøye, ofte i samarbeid med en revmatologitjeneste . Det har vært pågående diskusjon om bruk av ikke-steroide immunsuppressive midler som førstelinjebehandling for VKH sykdom. En nylig studie viste imidlertid ingen forskjeller i utfall mellom tidlig førstelinje immunmodulerende behandling (IMT) og prednisonbehandling alene . Videre er immunosuppressive og biologiske terapier dyre og krever nøye forbehandlingsevaluering samt hyppig oppfølging med blodarbeid for å vurdere alvorlige bivirkninger.
i kronisk tilbakevendende stadium kan hyppig residiv foreslå resistens mot kortikosteroidbehandling og foreslå behov for steroidsparende immunmodulerende behandling . Det foretrukne middel for steroidresistent tilbakefall eller steroidintoleranse er ciklosporin . Infliksimab, rituksimab, adalimumab og interferon alfa-2a er biologiske midler som også har blitt brukt til å behandle refraktær uveitt ved VKH-sykdom.
for å behandle fremre uveitt som ofte er forbundet med akutt VKH, bør topikale steroider (f.eks. prednisolonacetat 1%) og topisk cyloplegi (f. eks. cyklopentolat 1% eller atropin 1%) foreskrives avhengig av graden av betennelse i fremre kammer.
Okulære komplikasjoner er ofte forbundet med VKH sykdom. Gitt flere stadier og ulike presentasjoner der en pasient kan presentere MED VKH, kan behandlingen bli forsinket i mange tilfeller. I alvorlige FORMER FOR VKH og ved tilbakefall kan intraokulær betennelse være vanskelig å kontrollere og kan føre til strukturell skade. Over 50% av pasientene utvikler relaterte komplikasjoner, inkludert katarakt, sekundær glaukom, koroidale neovaskulære membraner, subretinal fibrose eller en kombinasjon av disse (Figur 6) .
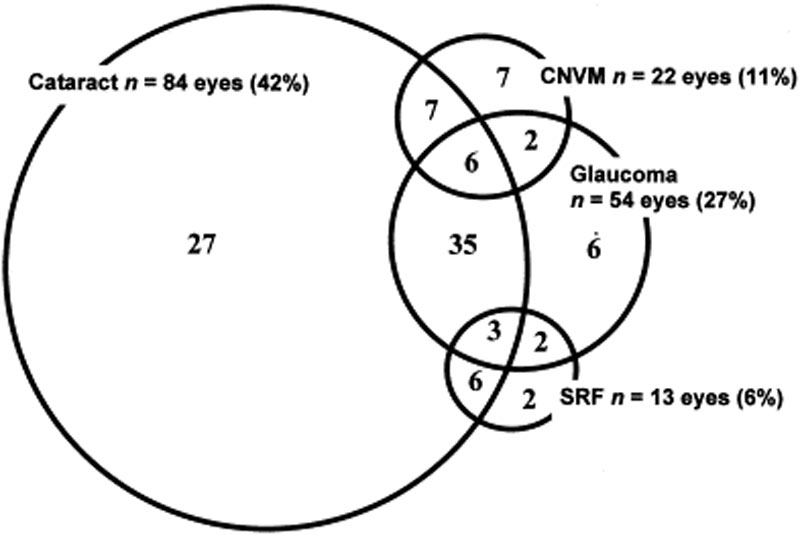
Figur 6: Venn-diagram som viser komplikasjoner hos VKH-pasienter. (Brukes med tillatelse Fra Am J Ophthalmol. 2001;131(5):599-606 )
EPIDEMIOLOGI og ETIOLOGI
- Typisk begynnelsesalder: 20 til 50 år
- Kvinnelig >mannlig
- Predisposisjon i populasjoner med pigmentert hud, spesielt Asiatisk, midtøsten, spanske og indianere
- genetiske risikoer: HLA-DR1, HLA-DR4, HLA-DRB1*0405 alleles
- Possible autoimmune targeting of melanocyte antigens
- Possible viral molecular mimicry as inciting factor in autoinflammatory activation
SIGNS
- Prodromal: Pleocytosis in CSF, meningitis, vertigo
- Uveitic: Bilateral granulomatous anterior or multifocal uveitis, multifocal serous retinal detachments in both eyes, optic nerve head edema and hyperemia
- Convalescent: Alopesi, poliose, solnedgang glød fundus, Sugiura tegn (limbal depigmentering), Dalen-Fuchs knuter under RPE
- Kronisk tilbakevendende: Tilbakefall av fremre uveitt, subretinal fibrose, koroidale neovaskulære membraner, multifokale chorioretinale arr, sekundær glaukom, grå stær
SYMPTOMER
- Influensalignende prodrom med hodepine, feber, sykdomsfølelse, kvalme og/eller oppkast
- Hørselstap, tinnitus og dysacusis
- nakkestivhet
- raskt forverret uklart syn som involverer begge øynene
- øye rødhet, smerte, flytere og/eller fotofobi
/li>
behandling/behandling
- høydose systemic corticosteroids (intravenous vs oral) for the acute phase with slow taper
- Immunosuppressive agents (cyclosporine) and biologics (infliximab) for recurrent disease refractory to corticosteroids
- Topical steroids (prednisolone acetate 1%) and cycloplegics (cyclopentolate 1% or atropine 1%) for anterior uveitis and ciliary spasm
- Intravitreal triamcinolone and/or intravitreal anti-VEGF injections for control of inflammation, choroidal neovascular membrane, and/or persistent subfoveal fluid
- Du L, Kijlstra A, Yang P. Vogt-Koyanagi-Harada sykdom: Ny innsikt i patofysiologi, diagnose og behandling. Prog Retin Øye Res 2016; 52: 84-111. https://PubMed.gov/26875727. DOI: 10.1016 / j. preteyeres.2016.02.002
- Yamaki K, Gocho K, Hayakawa K, Kondo I, Sakuragi S. Tyrosinase familieproteiner er antigener som er spesifikke for Vogt-Koyanagi-Harada sykdom. J Immunol 2000;165 (12): 7323-7329. https://PubMed.gov/11120868
- Horie Y, Takemoto Y, Miyazaki A, Namba K, Kase S, Yoshida K, Ota M, Hasumi Y, Inoko H, Mizuki N, Ohno S. Tyrosinase gen familie Og Vogt-Koyanagi-Harada sykdom Hos Japanske pasienter. Mol Vis 2006;12:1601-1605. https://PubMed.gov/17200659
- Ng JY, Luk FO, Lai TY, Pang CP. Påvirkning av molekylær genetikk I Vogt-Koyanagi-Harada sykdom. J Oftalmisk Inflamm Infisere 2014; 4: 20. https://PubMed.gov/25097674. DOI: 10.1186 / s12348-014-0020-1
- Bowling B. Uveitt. Kanski ‘ S Klinisk Oftalmologi New York, New York: Elsevier; 2016; kapittel 11; p. 395-465.
- YEH PT YC, Yang CH, Lin CP. Nonrhegmatogen Retinal Detachment. I: Schachat AP SS, Hinton DR, Wilkinson CP, Wiedemann P,, redaktør. Ryans Netthinnen. New York: Elsevier; 2018; kapittel 99; p. 1828-1849.
- Goto H RK, Rao N. Vogt-Koyanagi-Harada Sykdom. In: Schachat AP SS, Hinton DR, Wilkinson CP, Wiedemann P, redaktør. Ryans Netthinnen. New York, New York: Elsevier; 2018; kapittel 78; s. 1505-1515.Riddington L, Hall AJ, Tait B, Nicholson I, Varney M. Vogt-Koyanagi-Harada syndrom hos Pasienter Av Vietnamesisk herkomst. Aust N Z J Oftalmol 1996; 24 (2): 147-149. https://PubMed.gov/9199747
- Sugita S, Takase H, Kawaguchi T, Taguchi C, Mochizuki M. Kryssreaksjon mellom tyrosinase peptider og cytomegalovirus antigen Av T-celler fra pasienter Med Vogt-Koyanagi-Harada sykdom. Int Oftalmol 2007;27 (2-3): 87-95. https://PubMed.gov/17253112. DOI: 10.1007 / s10792-006-9020-y
- Freund BK SD, Mieler WF, Yannuzzi LA. Inflammasjon. Netthinneatlaset. New York, New York: Elsevier 2017; kapittel 4; p. 279-398.
- Rao N. Vogt-Koyanagi-Harada Sykdom. In: j YMaD, redaktør. Oftalmologi. New York, New York: Elsevier; 2014; kapittel 7.17; p.761-763.
- Rao NA, Xu S, Font RL. Sympatisk oftalmia. En immunhistokjemisk studie av epitelioid og gigantiske celler. Oftalmologi 1985; 92 (12): 1660-1662. https://PubMed.gov/4088616
- Nussenblatt RB. Vogt-Koyanagi-Harada Syndrom. In: Norsk journalist trakasserad, editor. Uveitt: Grunnleggende Og Klinisk Praksis. 4. Utgave ed: Elsevier; 2010; kapittel Kapittel 24.Les RW, Holland GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L, Pivetti-Pezzi P, Tessler HH, Usui M. Reviderte diagnostiske kriterier For Vogt-Koyanagi-Harada sykdom: rapport fra en internasjonal nomenklaturkomite. Er J Oftalmol 2001; 131 (5): 647-652. https://PubMed.gov/11336942
- Chung H, Choi DG. Klinisk analyse av uveitt. Koreansk J Oftalmol 1989; 3 (1): 33-37. https://PubMed.gov/2795939. DOI: 10.3341 / kjo.1989.3.1.33
- Abu El-Asrar AM, Al-Kharashi AS, Aldibhi H, Al-Fraykh H, Kangave D. Vogt-Koyanagi-Harada sykdom hos barn. Eye (Lond)) 2008;22(9):1124-1131. https://PubMed.gov/17479116. DOI: 10.1038 / sj.øyne.6702859
- Martin TD, Rathinam SR, Cunningham ET. Prevalens, kliniske egenskaper og årsaker til synstap hos barn Med Vogt-Koyanagi-Harada sykdom I Sør-India. Netthinnen 2010; 30 (7): 1113-1121. https://PubMed.gov/20168275. DOI: 10.1097 / IAE.0b013e3181c96a87
- Forster DJ, Grønn RL, Rao NA. Unilateral manifestasjon Av Vogt-Koyanagi-Harada syndromet i et 7 år gammelt barn. Am J Oftalmol 1991; 111 (3): 380-382. https://PubMed.gov/2000916
- Yamamoto Y, Fukushima A, Nishino K, Koura Y, Komatsu T, Ueno H. Vogt-koyanagi-harada sykdom med utbrudd hos eldre pasienter i alderen 68 til 89 år. Jpn J Oftalmol 2007; 51 (1): 60-63. https://PubMed.gov/17295144. DOI: 10.1007 / s10384-006-0379-0
- Wang Y, Chan CC. Kjønnsforskjeller i vogt-koyanagi-harada sykdom og sympatisk ophthalmia. J Oftalmol 2014; 2014: 157803. https://PubMed.gov/24734166. DOI: 10.1155/2014/157803
- Nakao K, Abematsu N, Mizushima Y, Sakamoto T. Optisk plate hevelse I Vogt-Koyanagi-Harada sykdom. Invester Oftalmol Vis Sci 2012; 53 (4): 1917-1922. https://PubMed.gov/22408010. DOI: 10.1167 / iovs.11-8984
- Rao NA, Gupta A, Dustin L, Chee SP, Okada AA, Khairallah M, Bodaghi B, Lehoang P, Accorinti M, Mochizuki M, Prabriputaloong T, Lese RW. Frekvens for å skille mellom kliniske egenskaper i Vogt-Koyanagi-Harada sykdom. Ophthalmology 2010;117(3):591-599, 599.e591. https://PubMed.gov/20036008. DOI: 10.1016/j.ophtha.2009.08.030
- Veerappan M, Fleischman D, Ulrich JN, Stinnett SS, Jaffe GJ, Allingham RR. The Relationship of Vogt-Koyanagi-Harada Syndrome to Ocular Hypertension and Glaucoma. Ocul Immunol Inflamm 2017;25(6):748-752. https://PubMed.gov/27438521. DOI: 10.1080/09273948.2016.1189578
- Baltmr A, Lightman S, Tomkins-Netzer O. Vogt-Koyanagi-Harada syndrome – current perspectives. Clin Ophthalmol 2016;10:2345-2361. https://PubMed.gov/27932857. DOI: 10.2147/OPTH.S94866
- Kitaichi N, Matoba H, Ohno S. den positive rollen av lumbal punktering i diagnosen Vogt-Koyanagi-Harada sykdom: lymfocyttundergrupper i vandig humor og cerebrospinalvæske. Int Oftalmol 2007;27 (2-3): 97-103. https://PubMed.gov/17211585. DOI: 10.1007 / s10792-006-9016-7
- Oshima Y, Harino S, Hara Y, Tano Y. Indocyanin grønne angiografiske funn I Vogt-Koyanagi-Harada sykdom. Am J Oftalmol 1996; 122 (1): 58-66. https://PubMed.gov/8659599
- Les RW, Yu F, Accorinti M, Bodaghi B, Chee SP, Fardeau C, Goto H, Holland Gn, Kawashima H, Kojima E, Lehoang P, Lemaitre C, Okada AA, Pivetti-Pezzi P, Secchi A, Se RF, Tabbara KF, Usui M, Rao NA. Evaluering av effekten på utfall av administreringsveien for kortikosteroider ved akutt Vogt-Koyanagi-Harada sykdom. Er J Oftalmol 2006; 142 (1): 119-124. https://PubMed.gov/16815259. DOI: 10.1016 / j.ajo.2006.02.049
- Rubsamen PE, Gass JD. Vogt-Koyanagi-Harada syndrom. Klinisk kurs, terapi og langsiktig visuelt utfall. Arch Ophthalmol 1991;109(5):682-687. https://PubMed.gov/2025171
- Urzua CA, Velasquez V, Sabat P, Berger O, Ramirez S, Goecke A, Vá Dh, Gatica H, Guerrero J. Tidligere immunmodulerende behandling er forbundet med bedre visuelle resultater i en undergruppe av pasienter med Vogt-Koyanagi-Harada sykdom. Acta Oftalmol 2015; 93 (6):e475-480. https://PubMed.gov/25565265. DOI: 10.1111 / aos.12648
- Les RW, Rechodouni A, Butani N, Johnston R, LaBree LD, Smith RE, Rao NA. Komplikasjoner og prognostiske faktorer I Vogt-Koyanagi-Harada sykdom. Er J Oftalmol 2001; 131 (5): 599-606. https://PubMed.gov/11336934
Foreslått Referanseformat
Mai AP, Tran C, Wilson Cw, Fox AR, Boldt HC. Vogt-Koyanagi-Harada (VKH) Sykdom. EyeRounds.org. 1.April 2019. Tilgjengelig frahttp://EyeRounds.org/cases/284-vogt-koyanagi-harada.htm
sist oppdatert: 04/1/2019 - Tidlige manifestasjoner av sykdom.
Leave a Reply