Maladie de Vogt-Koyanagi-Harada (VKH)
Auteurs: Anthony P. Mai, BS; Charlene Tran, BS; Caroline W. Wilson, MD; Austin R. Fox, MD; H. Culver Boldt, MD
1er avril 2019
PRÉSENTATION INITIALE
Plainte principale
Vision floue et maux de tête
Antécédents de maladie actuelle
Une Vietnamienne de 44 ans s’est présentée au service des urgences avec des antécédents de vision floue progressive des deux yeux pendant 10 jours et des antécédents de maux de tête sévères pendant trois jours. Sa perte de vision centrale ne s’était pas améliorée avec une réfraction de son optométriste. Ses maux de tête occipitaux sévères s’aggravaient avec le mouvement et étaient associés à un malaise généralisé, une fatigue extrême, une photophobie légère et des déchirures. L’acétaminophène a partiellement atténué la douleur.
Elle s’était récemment rendue au Vietnam mais a nié y avoir rencontré des contacts malades. Elle a nié la claudication de la mâchoire, les fièvres ou les changements de poids. Elle a nié les éruptions cutanées, les changements auditifs, les acouphènes, les étourdissements, les engourdissements ou les picotements. Elle a nié avoir jamais eu la tuberculose. Elle n’avait aucun antécédent de problèmes de vision, de maladies auto-immunes ou de cancer.
Antécédents oculaires
- Antécédents de chirurgie esthétique des paupières (blépharoplastie bilatérale) trois ans auparavant
- Pas d’antécédents de traumatisme ou de maladie oculaire
Antécédents médicaux
Aucun
Médicaments
Acétaminophène au besoin
Allergies
Aucune allergie médicamenteuse connue
Antécédents familiaux
Pas d’antécédents de maladie oculaire ou de maladie auto-immune
Antécédents sociaux
Elle a immigré du Vietnam plusieurs années avant la présentation. Elle est mariée et a trois enfants. Elle travaille dans un salon de manucure. Elle ne consomme pas de produits du tabac, d’alcool ou de substances illicites. Elle voyage au Vietnam tous les six à douze mois.
Examen des systèmes
Négatif sauf pour ce qui est détaillé dans l’histoire de la maladie actuelle
EXAMEN OCULAIRE
Acuité visuelle avec / sans correction (Snellen)
- Œil droit (DO): 20/300 (aucune amélioration avec sténopé)
- Œil gauche (OS): 20/60-2+2 (pas d’amélioration avec sténopé)
Motilité oculaire / Alignement
Mouvements extraoculaires complets des deux yeux (OU)
Pression intraoculaire (PIO): (Tonopen)
- DO: 12 mmHg
- OS: 14 mmHg
Pupilles
- DO: 4 mm dans l’obscurité, 3 mm dans la lumière, pas de défaut pupillaire afférent relatif (RAPD)
- OS: 4 mm dans l’obscurité, 3 mm dans la lumière, pas de RAPD
Champs visuels de confrontation: (Compter ecotome central
Externe
Normal des deux côtés
Examen à la lampe à fente
- Paupières/cils : Normal OU
- Conjonctive/sclérotique : Claire et silencieuse OU
- Cornée: 1+ érosions épithéliales ponctuelles, pas de précipités kératiques OU
- Chambre antérieure: Cellule trace et évasement et profonde OU
- Iris: Architecture normale OU
- Lentille: Claire OU
Examen du fond d’œil dilaté (DFE)
- Vitré : Cellules vitrées antérieures traces OU
- Disque:
- OD: Œdème discal de grade 3 , hyperémique
- OS: Œdème discal de grade 2-3, hyperémique
- Rapport Coupe-disque: 0,0 OU
- Macula:
- OD: œdème maculaire cystoïde 3 + (EMC) et liquide sous-rétinien (SRF) s’étendant du disque à la macula temporale. Pas de lipides ni d’exsudats. Choroïde d’apparence marécageuse.
- OS: 2+ CME et SRF s’étendant du disque à travers la fovéa. 1-2+ lipide linéaire s’étendant du disque vers la fovéa. Choroïde d’apparence marécageuse.
- Vaisseaux:
- OD: Gainage temporel
- OS: Normal
- Périphérie:
- OD: Touffe rétinienne kystique antérieure à l’équateur à 10:30
- OS: SRF peu profonde antérieure à l’équateur à 4:00
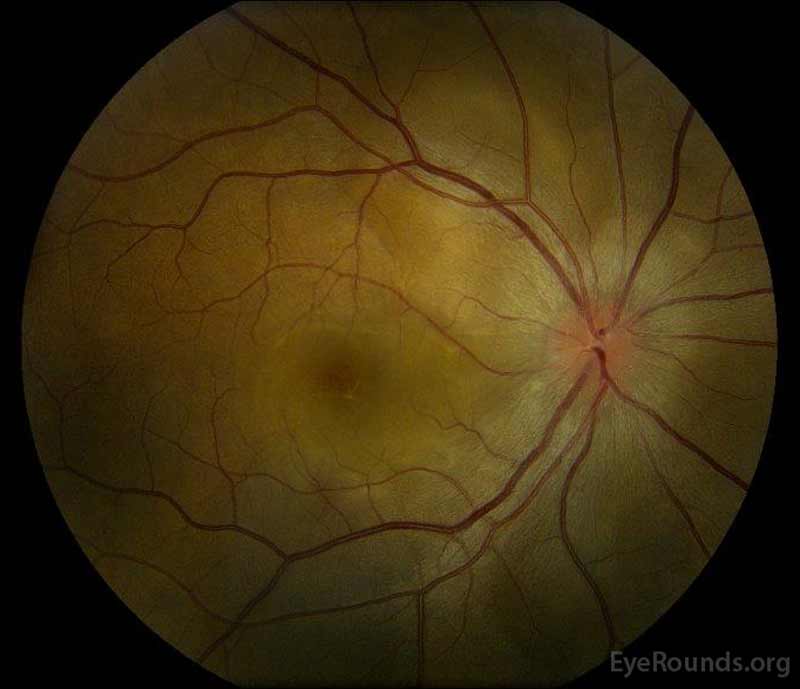 |
|
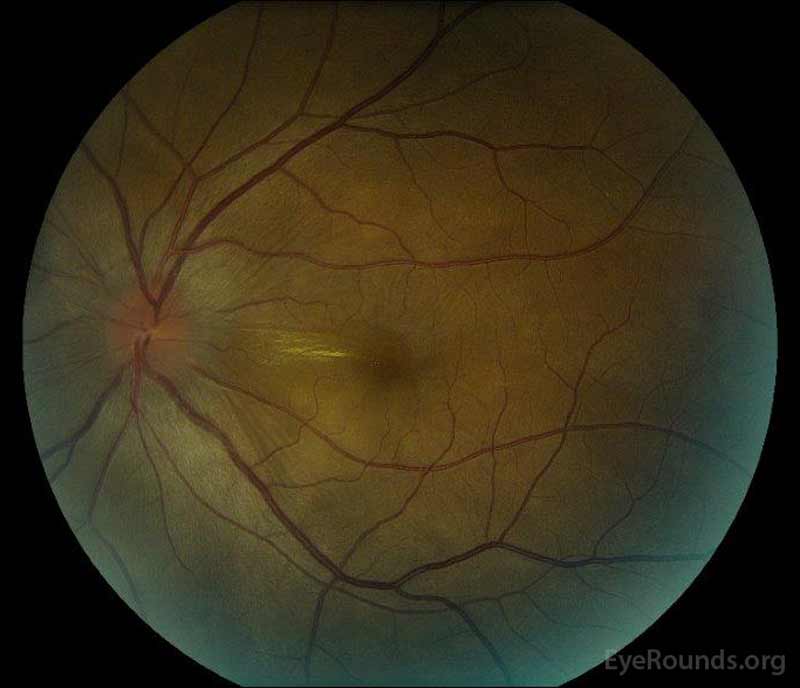
Figure 1: Photographies du fond d’œil en couleur lors de la présentation: (image de gauche) L’œil droit présente un œdème discal et une légère hyperémie ainsi qu’un liquide sous-rétinien s’étendant du disque temporellement à travers la macula. Il y a aussi un décollement séreux de la rétine focal superotemporal au disque, le long de l’arcade supérieure. (Right image) The left eye has disc edema and mild hyperemia, along with subretinal fluid extending from the disc to the macula and linear lipid deposits in the nasal macula.
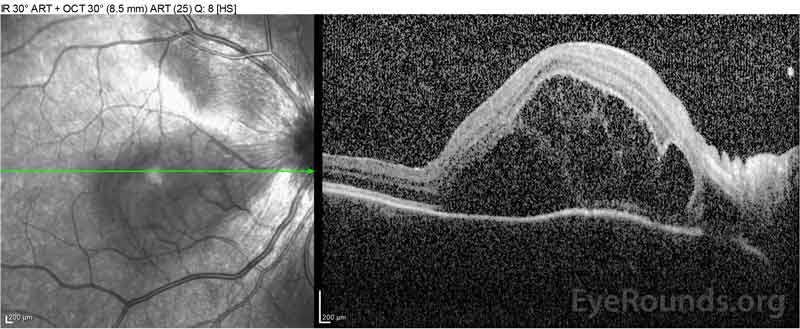 |
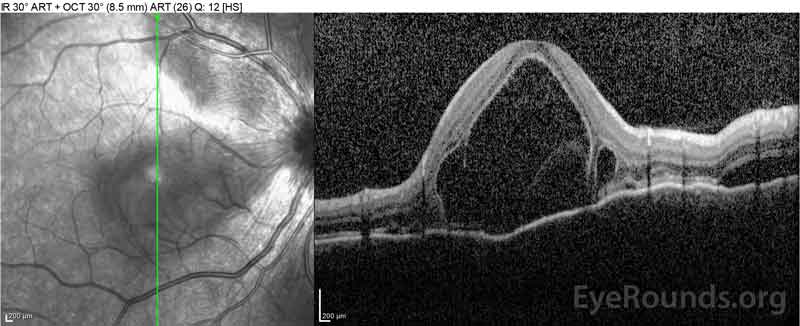 |
 |
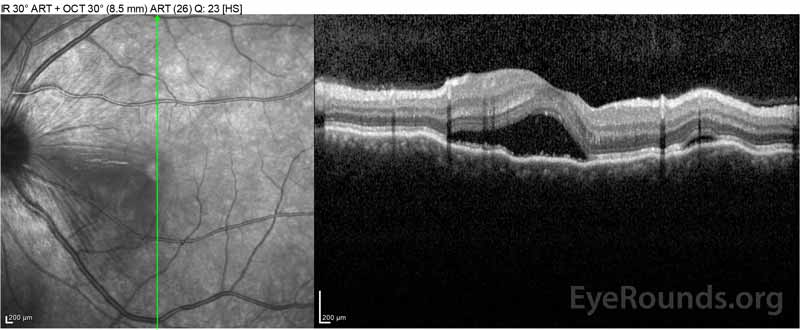 |
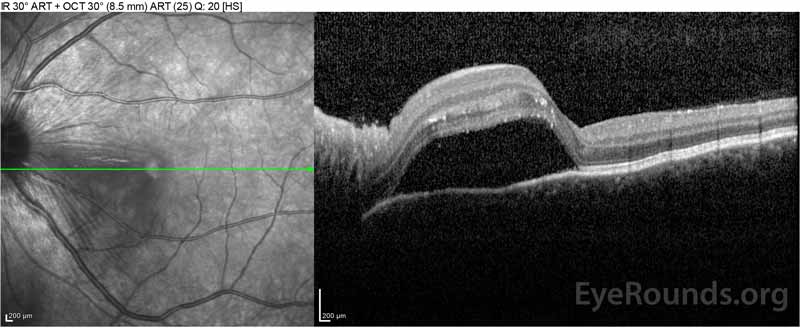
Figure 2: La tomographie par cohérence optique (OCT) de l’œil droit (panneaux supérieurs) montre un décollement séreux de la rétine impliquant la fovéa avec un liquide intra-rétinien sus-jacent étendu, une perturbation des couches rétiniennes externes et des ondulations de la choroïde épaissie. L’OCT de l’œil gauche (panneaux inférieurs) montre un décollement séreux de la rétine dans la macula nasale s’étendant jusqu’à la fovéa.
 |
|
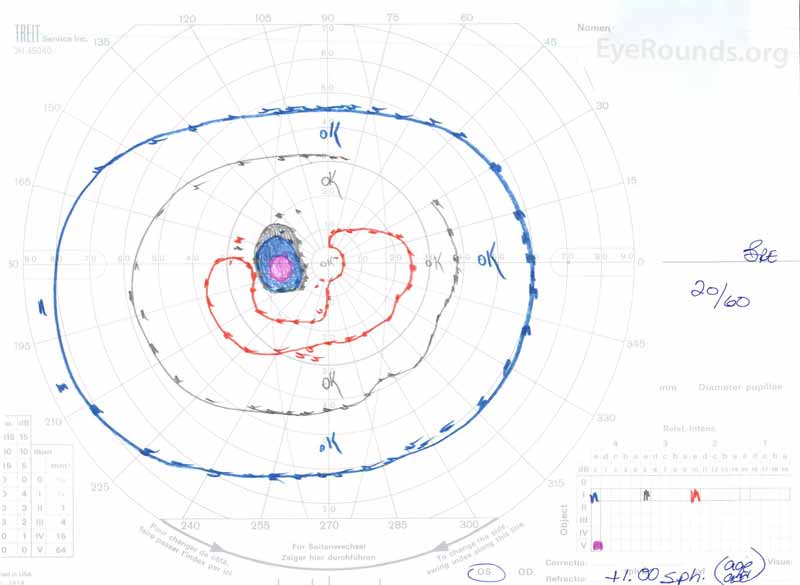
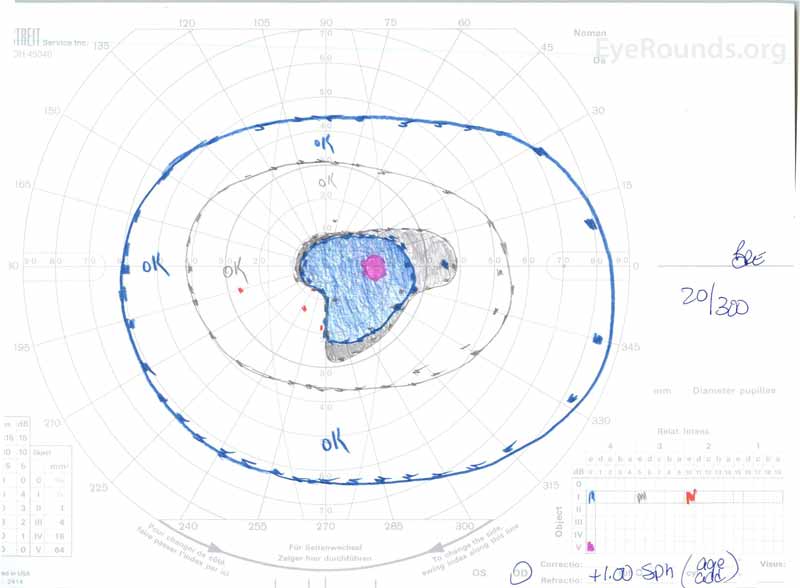
Figure 3: Champs visuels Goldman (GVF), OU. (Image de gauche) L’OS montre une tache aveugle physiologique élargie et un scotome central léger. (Image de droite) OD montre un scotome central moyennement dense incorporant l’angle mort physiologique et s’étendant de manière inférotemporelle.
B-scan: Aucun signe de sclérite, opacités vitréennes légères / cellules inférieures
Diagnostic différentiel
- Épithéliopathie pigmentaire placoïde multifocale postérieure aiguë (APMPPE)
- choriorétinopathie séreuse centrale
- Névrite optique
- Panuvéite
- Maladie auto-immune (par exemple, LED, sarcoïdose)
- Infection (e.g. , syphilis, tuberculose, Bartonella henselae)
- Malignité (par exemple, lymphome oculaire)
- Sclérite postérieure
- Ophtalmie sympathique
- Syndrome d’épanchement uvéal
- Syndrome de Vogt-Koyanagi-Harada
BILAN sanguin
Numération formule sanguine complète
Nombre de globules blancs: 4,9 K / mm3 (Réf: 3,7-10,5)
Nombre de globules rouges 3,99 M / mm3 (Réf: 4,0-5,2)
Hémoglobine 11,6 g / dL (Réf: 11,9-15,5)
Hématocrite 35% (Réf:: 35-47)
Basic metabolic panel
Sodium 138 mEq/L (Ref: 135-145)
Potassium 4.3 mEq/L (Ref: 3.5-5.0)
Chloride 107 mEq/L (Ref: 95-107)
CO2 20 mEq/L (Ref: 22-29)
Blood urea nitrogen 16 mEq/dL (Ref: 10-20)
Creatinine 0.7 mg/dL (Ref: 0.5-1.0)
C-reactive Protein (CRP): <0.5 mg/dL (Ref: <=0.5)
Erythrocyte sedimentation rate (ESR): 12 mm/Hr (Ref: 0-20)
Angiotensin–converting enzyme (ACE): 13 U/L (Ref: 8-52)
QuantiFERON-TB Gold: négatif
Fer, sang 54 microgrammes / dL (Réf:37-145)
Capacité totale de liaison au fer 379 microgrammes / dL (Réf: 250-425)
ÉVOLUTION CLINIQUE
La patiente a d’abord été évaluée par le service des urgences en raison de ses plaintes de maux de tête sévères à nouveau apparus et de perte de vision. La tomodensitométrie cérébrale (TDM) et l’imagerie par résonance magnétique (IRM) n’étaient pas remarquables. La VS et la CRP se situaient dans les niveaux normaux. La clinique d’ophtalmologie l’a évaluée le lendemain et a trouvé des détachements rétiniens séreux bilatéraux et une panuvéite. ACE et QuantiFERON-TB Gold labs étaient tous deux négatifs. Elle a reçu un diagnostic de maladie de Vogt-Koyanagi-Harada en raison de sa présentation clinique et de son origine asiatique. Elle a été traitée avec 80 mg de prednisone par jour, de l’acétaminophène au besoin pour les maux de tête et une supplémentation en vitamine D et en calcium. Ses maux de tête se sont rapidement résolus et son acuité visuelle s’est régulièrement améliorée au cours des deux semaines suivantes. Sa posologie de prednisone a ensuite été réduite à 40 mg sur trois semaines avec une résolution continue des symptômes et une amélioration de l’acuité visuelle. Elle n’avait pas de récurrence de maux de tête ou d’aggravation de la vision pendant la conicité de la prednisone. Lors de son dernier rendez-vous, elle avait diminué jusqu’à 5 mg tous les deux jours, sans retour des symptômes. Son acuité visuelle lors de cette visite de suivi était de 20/15-2 DO et de 20/20 + 2 OS, et l’OCT maculaire a montré une résolution complète de l’œdème discal et des détachements rétiniens séreux dans les deux yeux (Figure 4).
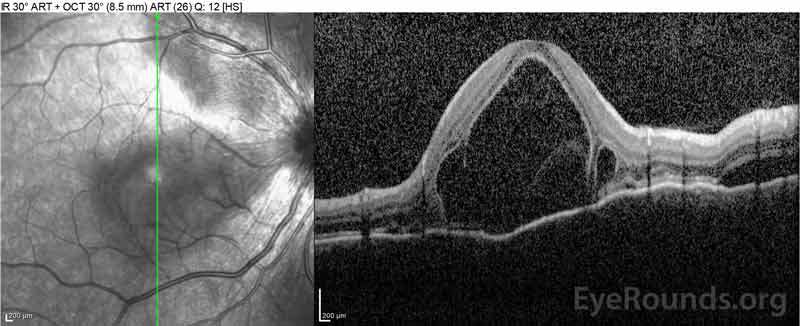
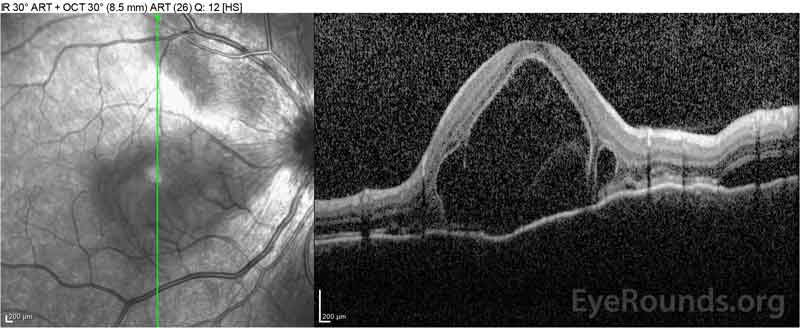
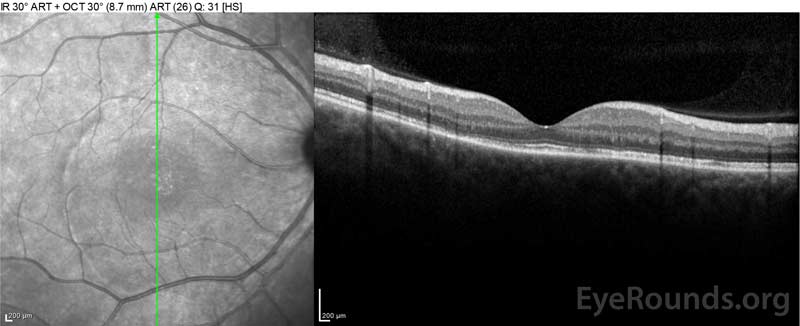
Figure 4: Tomographie par cohérence optique montrant le liquide sous-rétinien au début (en haut) et le cours de la résolution à une semaine (au milieu) et à cinq semaines (en bas) lors d’une conicité de prednisone orale à forte dose. Notez le lissage des ondulations choroïdiennes avec traitement.
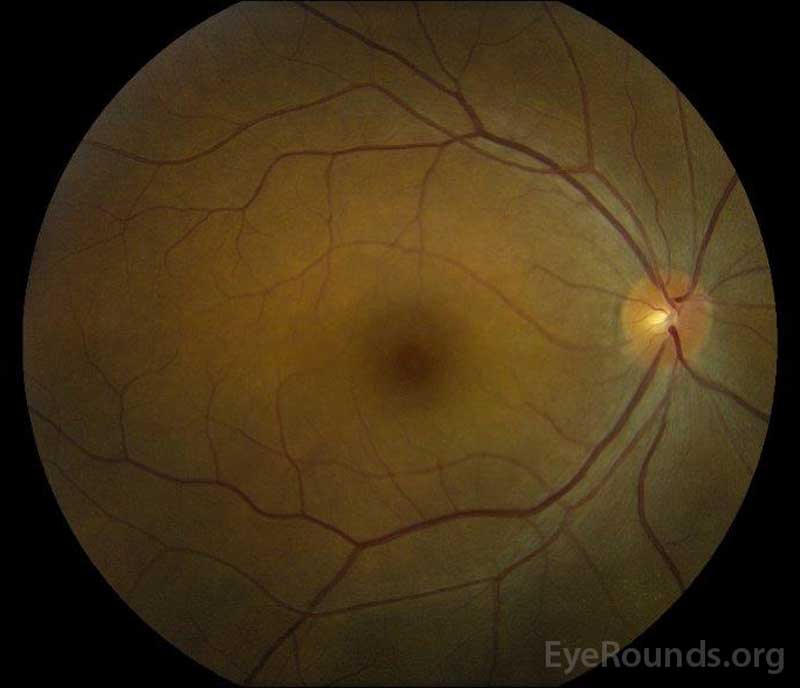 |
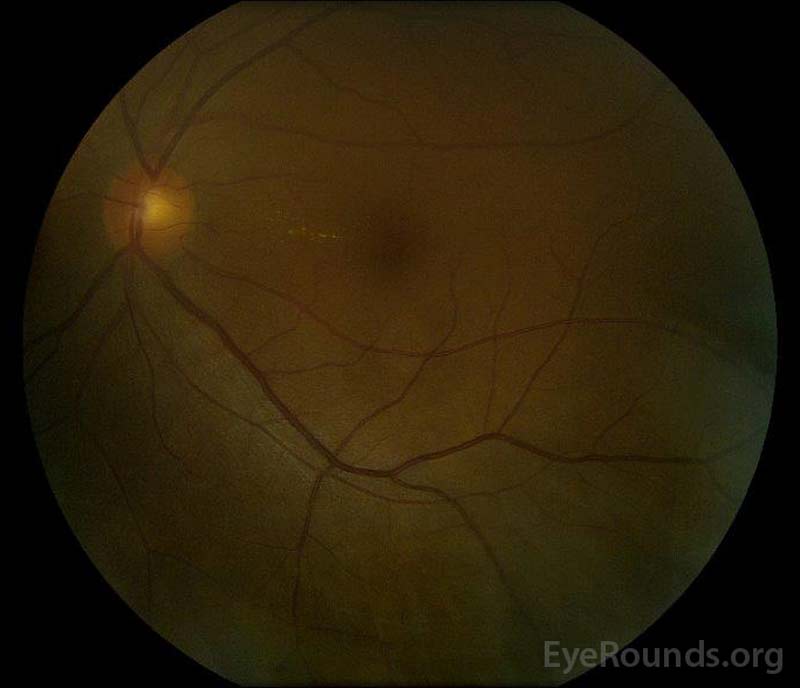
|
Figure 5: Photographie du fond d’œil en couleur des yeux droit (A) et gauche (B) pendant la phase de convalescence démontrant une amélioration du liquide sous-rétinien et de l’œdème discal.
DIAGNOSTIC
Maladie incomplète de Vogt-Koyanagi-Harada
DISCUSSION
La maladie de Vogt-Koyanagi-Harada (VKH) est une maladie auto-immune systémique caractérisée par une panuvéite granulomateuse bilatérale non nécrosante associée à des modifications tégumentaires extraoculaires, telles que la poliose et le vitiligo, et une inflammation affectant l’uvée, l’oreille interne, les cheveux et les méninges. La maladie de Harada est l’uvéite isolée sans les signes ou symptômes systémiques de la VKH.
Étiologie
L’étiologie de la maladie VKH est encore largement inconnue malgré les efforts de recherche actuels. On pense qu’il s’agit d’une maladie auto-immune acquise impliquant une hypersensibilité médiée par les cellules T aux auto-antigènes mélanocytaires, avec une prédisposition génétique sous-jacente et un déclencheur microbien possible. La tyrosinase et les peptides liés à la tyrosinase sont des antigènes de mélanocytes qui ont été suggérés comme cibles des processus auto-immuns dans VKH. Cependant, un risque accru de maladie VKH n’était pas associé à la famille des gènes de la tyrosinase, selon une étude.
En raison de la prévalence accrue parmi certains groupes ethniques et de genre, on pense qu’il y a une prédisposition génétique dans la pathogenèse de la VKH. Plusieurs gènes, y compris les gènes de l’antigène leucocytaire humain (HLA) et de l’interleukine (IL), ont été associés à la VKH dans différentes populations ethniques. Les récepteurs HLA sont des complexes majeurs d’histocompatibilité chez l’homme qui présentent des peptides au système immunitaire. HLA-DR1, HLA-DR4, HLA-DRB1 * 0405 et HLA-DRw53 sont plusieurs haplotypes trouvés chez des patients atteints de la maladie VKH. HLA-DR4 est plus fréquent chez les Japonais et les Hispaniques, tandis que HLA-DRB1 * 0405 est plus fréquent chez les patients coréens et du Moyen-Orient. Les allèles HLA-DR4 et HLA-DRB1 * 0405 sont trouvés chez les patients vietnamiens. Le récepteur HLA-DRB1 se lie aux antigènes des mélanocytes à différentes capacités. Malgré ces associations, les tests génétiques ne sont pas recommandés pour le moment.
Compte tenu des symptômes prodromiques habituels qui accompagnent l’HVK, notamment fièvre, maux de tête, méningisme et acouphènes, une étiologie virale incitative a été suggérée comme déclencheur de l’apparition de l’HVK par des mécanismes de mimétisme moléculaire chez des patients génétiquement prédisposés. La glycoprotéine H de l’enveloppe du cytomégalovirus présente une homologie significative des acides aminés avec le peptide tyrosinase, et l’infection à CMV peut déclencher la VKH par mimétisme moléculaire (c.-à-d. reconnaissance par les récepteurs de classe II HLA). Le virus Ebstein-bar (EBV) a également été impliqué. Cependant, il n’y a pas eu de preuve définitive concernant une étiologie virale de la VKH, et on ne sait toujours pas ce qui déclenche la réponse auto-immune de la VKH.
Physiopathologie
Il existe quatre phases classiques de VKH qui peuvent avoir des présentations variables: prodromique, uvéitique aiguë, convalescente et chronique-récurrente. Les changements histopathologiques commencent généralement dans la phase aiguë.
La phase uvéitique aiguë est caractérisée par un épaississement uvéal bilatéral secondaire à une inflammation granulomateuse. Les granulomes sont constitués de lymphocytes, de macrophages et de cellules épithélioïdes et géantes remplies de granulés. Bien que les cellules épithélioïdes étaient auparavant considérées comme des mélanocytes altérés, une étude immunohistochimique de suivi a suggéré une origine des macrophages tissulaires à la place. Des granulomes remplis d’histiocytes épithélioïdes, appelés nodules de Dalen-Fuchs, peuvent souvent être observés entre l’épithélium pigmentaire rétinien (EPR) et la membrane de Bruch. L’inflammation granulomateuse uvéale entraîne un épaississement choroïdien et des détachements rétiniens exsudatifs remplis de liquide protéique. De plus, la pléocytose (i.e., augmentation du nombre de cellules) peut être présent dans la chambre antérieure et vitré.
La phase de convalescence est identifiée par dépigmentation de la choroïde et des zones extraoculaires, y compris la peau périoculaire et les cheveux. Une choroïde dépigmentée posée contre un nerf optique pâle donne l’impression d’un fond d’œil « à la lueur du coucher du soleil », ce qui est une caractéristique classique de cette phase de VKH. De plus, les nodules de Dalen-Fuchs deviennent plus proéminents sous l’EPR pendant la phase de convalescence.
La phase chronique récurrente est caractérisée par une diminution de l’épaisseur choroïdienne, la résolution des détachements rétiniens séreux, une vitrite chronique légère et une inflammation récurrente du segment antérieur granulomateux. La néovascularisation choroïdienne (CNV) et la fibrose sous-rétinienne peuvent se développer au cours de cette phase et sont des indicateurs de progression grave de la maladie. La cataracte et le glaucome secondaire sont d’autres complications d’une inflammation de longue date ou récurrente dans cette phase.
Épidémiologie
La VKH est répandue chez les races à pigment cutané plus foncé, en particulier les Asiatiques, les Sud-Américains, les Moyen-Orientaux et les Amérindiens. La maladie VKH représente >10% des uvéites dans ces populations. Seulement 1 à 4% des cas d’uvéite seraient secondaires à la maladie VKH aux États-Unis (7). Aux États-Unis, la plupart des cas de VKH ont été trouvés pour affecter des individus d’origine asiatique, hispanique et / ou amérindienne décente. Fait intéressant, la maladie VKH affecte rarement les Africains malgré leur pigmentation sombre. L’incidence de la maladie VKH varie considérablement d’un sous-groupe racial à l’autre dans les pays voisins. Par exemple, l’incidence de la VKH en Corée n’est que de 2%, bien inférieure à celle observée au Japon et en Chine.
L’HVK a un début typique de 20 à 50 ans; cependant, des études suggèrent que 3,1 à 13,4% des cas d’HVK sont des patients pédiatriques et que 10% des cas ont ≥65 ans. Classiquement, on pense que VKH a une prédilection pour le sexe féminin, et bien que la plupart des études montrent que VKH affecte de manière disproportionnée les femmes, quelques études ont montré une prédisposition masculine ou aucune prédisposition sexuelle.
Signes / symptômes
Comme mentionné ci-dessus, les quatre stades de la maladie VKH sont prodromiques, uvéitiques, convalescentes et chroniques récurrentes. Chaque étape présente des caractéristiques cliniques distinctes.
- Prodromique: Ce stade initial peut se présenter comme une maladie grippale avec des symptômes principalement constitutionnels, tels que des maux de tête, des étourdissements, de la fièvre, de la fatigue et / ou des nausées. Des symptômes neurologiques de méningite, de paralysie du nerf crânien et de névrite optique, ainsi que des symptômes auditifs d’acouphènes, de dysacousie et de vertiges ont été rapportés. La photophobie, la vision floue, les flottements et / ou la douleur oculaire commencent généralement dans les 48 heures suivant les symptômes prodromiques. La phase prodromique dure généralement de quelques jours à quelques semaines.
- Uvéite aiguë: Cette étape comprend une vision floue, une photophobie, une injection conjonctivale et une douleur oculaire. Il peut y avoir une légère uvéite antérieure qui semble d’abord non granulomateuse. L’apparition unilatérale passe généralement à une implication bilatérale dans les 1 à 2 semaines. Une uvéite antérieure granulomateuse avec des précipités kératiques gras de mouton peut se développer. Les résultats de l’examen postérieur peuvent inclure un œdème et une hyperémie du nerf optique, des zones multifocales de choroïdite, de multiples zones de détachements rétiniens séreux localisés au fond d’œil postérieur, un épaississement choroïdien, des plis choriorétiniens rayonnants et une vitrite. Les détachements rétiniens séreux peuvent former un motif de trèfle dans le fond d’œil postérieur et peuvent évoluer vers de vastes détachements bulleux dans les cas graves. Le glaucome inflammatoire aigu a été associé à cette phase de la maladie et peut se présenter avec une chambre antérieure peu profonde secondaire à un œdème du corps ciliaire, imitant la fermeture à angle aigu. La durée de la phase uvéitique aiguë dépend du diagnostic et de la prise en charge rapides.
- Uvéite chronique ou Convalescente: Ce stade se développe généralement plusieurs semaines après la phase aiguë et se caractérise par le vitiligo (par exemple, le visage, les mains, les épaules ou le dos), la poliose et l’alopécie. Une dépigmentation près du limbe cornéen, connue sous le nom de signe de Sugiura, peut être observée un mois après l’apparition de la maladie; cependant, ce signe est rarement observé en dehors de la population japonaise. La dépigmentation choroïdale se produit généralement sur quelques mois et donne la couleur rouge orangé vif de la choroïde et du fond d’œil classique « sunset glow ». »Le fond d’œil Sunset glow est considéré comme le plus important et le plus prédictif dans le diagnostic de l’HVK chronique. Des cicatrices choriorétiniennes bien définies, rondes et nummulaires peuvent se former à la périphérie médiane. La phase uvéitique chronique dure généralement plusieurs mois.
- Chronique – récurrente: Ce stade est caractérisé par des épisodes récurrents d’uvéite antérieure granulomateuse avec précipités kératiques de graisse de mouton, nodules d’iris, dépigmentation de l’iris, synéchies postérieures, cataractes sous-capsulaires postérieures, glaucome secondaire, membranes néovasculaires choroïdiennes et, finalement, fibrose sous-rétinienne et atrophie choriorétinienne nummulaire. La phase chronique se développe généralement au moins six mois après la présentation initiale. Les détachements rétiniens séreux présents pendant les phases aiguë et de convalescence ne se reproduisent généralement pas avec un traitement corticostéroïde agressif.
Critères de diagnostic
Les critères de diagnostic les plus récents, nommés Critères de Diagnostic révisés (RDC) pour l’HVK, ont été définis en 1999 lors du Premier Atelier international sur l’HVK. Celles-ci sont décrites dans le tableau 1. Les CDR sont utiles en ce qu’ils divisent les VKH en trois catégories diagnostiques différentes en fonction de la phase de la maladie au cours de laquelle un patient se présente: complète, incomplète et probable. Cette catégorisation de la maladie permet une prise en charge appropriée et précoce de la maladie « probable » qui peut aider à prévenir la progression vers une maladie « complète ».
Il est essentiel de rechercher d’autres causes d’inflammation oculaire, infectieuses et auto-inflammatoires. Ceux-ci peuvent inclure la vitesse de sédimentation des érythrocytes (ESR), la protéine C-réactive (CRP), le test quantiferon-Gold pour la tuberculose, la réactif plasmatique rapide (RPR) pour la syphilis, l’enzyme de conversion de l’angiotensine (ACE) et une radiographie pulmonaire pour la sarcoïdose, les anticorps antinucléaires (ANA) et p- / c-ANCA. En outre, des antécédents de traumatisme oculaire récent ou de chirurgie intraoculaire doivent être notés et suggèrent probablement une ophtalmie sympathique (SO) comme diagnostic le plus probable étant donné la présentation et la physiopathologie très similaires partagées entre SO et VKH.
Pour soutenir un diagnostic de VKH dans des cas équivoques, une ponction lombaire peut être réalisée pour rechercher une pléocytose lymphocytaire et monocytaire; cependant, cela est rarement utilisé cliniquement. Quatre-vingt pour cent des patients ont une pléocytose dans le liquide céphalo-rachidien (LCR) en une semaine et 97% ont une pléocytose en trois semaines. L’augmentation des niveaux de cellules immunitaires peut durer jusqu’à huit semaines après l’apparition de la maladie. Les profils de marqueurs de surface des lymphocytes T sont similaires entre le LCR et l’humeur aqueuse, mais différents du sang. Cela suggère la capacité du LCR à refléter avec précision l’inflammation uvéale dans la maladie VKH.
Tableau 1. Critères diagnostiques révisés pour la maladie de Vogt-Koyanagi-Harada
* Du tableau 1 de (15).
« Maladie de Vogt-Koyanagi-Harada complète (les critères 1 à 5 doivent être présents)
- Aucun antécédent de traumatisme oculaire pénétrant ou de chirurgie précédant l’apparition initiale de l’uvéite.
- Aucune preuve clinique ou de laboratoire suggérant d’autres entités de la maladie oculaire.
- Atteinte oculaire bilatérale (a ou b doivent être atteints, selon le stade de la maladie lors de l’examen du patient).
- Manifestations précoces de la maladie.
- Il doit y avoir évidence d’une choroïdite diffuse (avec ou sans uvéite antérieure, réaction inflammatoire vitréenne ou hyperémie du disque optique), qui peut se manifester par l’une des zones focales du liquide sous-rétinien, ou par des détachements rétiniens séreux bulleux.
- Avec des résultats de fond d’œil équivoques; les deux éléments suivants doivent également être présents:
- Zones focales de retard de perfusion choroïdienne, zones multifocales de fuite ponctuelle, grandes zones placoïdes d’hyperfluorescence, regroupement dans le liquide sous-rétinien et coloration du nerf optique (listée par ordre d’apparition séquentielle) par angiographie à la fluorescéine et
- Épaississement choroïdien diffus, sans preuve de sclérite postérieure par échographie.
- Manifestations précoces de la maladie.
- Manifestations tardives de la maladie.
- Histoire suggérant la présence antérieure de résultats de 3a, et soit les deux (2) et (3) ci-dessous, soit plusieurs signes de (3):
- dépigmentation oculaire (l’une des manifestations suivantes est suffisante): (a) Fond d’œil à la lueur du coucher du soleil, ou (b) signe de Sugiura.
- Autres signes oculaires :
- Cicatrices dépigmentées choriorétiniennes nummulaires, ou
- agglutination et/ou migration de l’épithélium pigmentaire rétinien, ou
- uvéite antérieure récurrente ou chronique.
- Méningisme (malaise, fièvre, maux de tête, nausées, douleurs abdominales, raideur du cou et du dos, ou une combinaison de ces facteurs; les maux de tête seuls ne suffisent cependant pas à répondre à la définition du méningisme), ou
- Acouphènes, ou
- pléocytose du liquide céphalo-rachidien.
- Alopécie, ou
- Poliose, ou
- Vitiligo.
Maladie de Vogt-Koyanagi-Harada incomplète (les critères 1 à 3 et 4 ou 5 doivent être présents)
- Aucun antécédent de traumatisme oculaire pénétrant ou de chirurgie précédant l’apparition initiale de l’uvéite, et
- Aucune preuve clinique ou de laboratoire suggérant d’autres entités de la maladie oculaire, et
- Atteinte oculaire bilatérale.
- Résultats neurologiques / auditifs; tels que définis pour la maladie complète de Vogt-Koyanagi-Harada ci-dessus, ou
- Résultats tégumentaires; tels que définis pour la maladie complète de Vogt-Koyanagi-Harada ci-dessus.
Maladie probable de Vogt-Koyanagi-Harada (maladie oculaire isolée; les critères 1 à 3 doivent être présents)
- Aucun antécédent de traumatisme oculaire pénétrant ou de chirurgie précédant l’apparition initiale de l’uvéite.
- Aucune preuve clinique ou de laboratoire suggérant d’autres entités de la maladie oculaire.
- Atteinte oculaire bilatérale telle que définie pour la maladie complète de Vogt-Koyanagi-Harada ci-dessus. «
Tests / Bilan de laboratoire
Dans le bilan initial de VKH, il faut envisager d’obtenir les tests suivants:
- Tomographie par cohérence optique (OCT): Dans la phase uvéitique aiguë, l’OCT montrera probablement un épaississement choroïdien significatif et des détachements rétiniens séreux. Les accumulations de liquide sous-rétinien peuvent avoir des cloisons considérées comme des membranes de fibrine et des produits inflammatoires, créant une structure lobulaire qui peut également être vue sur l’angiographie à la fluorescéine. En phase de convalescence, l’OCT peut détecter des zones d’amincissement de la rétine après une inflammation résolue après un traitement aux corticostéroïdes.
- Échographie à balayage B: Dans la phase aiguë, l’échographie peut montrer un épaississement choroïdien postérieur diffus, un épaississement scléral postérieur, des détachements rétiniens et des opacités vitrées. Des épanchements ciliaires peuvent être observés par biomicroscopie échographique. Ce test est également utile pour éliminer la sclérite postérieure.
- Angiographie à la fluorescéine (FA): Classiquement, la FA révèle des points hypofluorescents choroïdiens multifocaux à la phase précoce suivis de multiples zones hyperfluorescentes focales avec fuite diffuse à la phase tardive. Le colorant fuit à travers l’EPR et s’accumule dans l’espace sous-rétinien entourant les points hyperfluorescents. FA peut être utile sur le plan diagnostique lorsque la maladie VKH se présente sans symptômes extraoculaires. Une hyperfluorescence du disque optique et des défauts de fenêtre causés par des cicatrices choriorétiniennes atrophiques peuvent être observés à la périphérie médiane. La FA au stade chronique-récurrent de la maladie VKH présente des défauts de fenêtre non spécifiques dus à des lésions de l’EPR, à une néovascularisation choroïdienne et à une fibrose sous-rétinienne.
- Angiographie au vert d’indocyanine (ICG): L’ICG de phase précoce représente des vaisseaux stromaux hyperfluorescents qui indiquent une vasculopathie choroïdienne et des points sombres hypofluorescents qui correspondent à des granulomes et à un remplissage inégal retardé du système vasculaire choroïdien. La phase tardive révèle des motifs vasculaires stromaux flous et une hyperfluorescence choroïdienne diffuse. L’hyperfluorescence discale suggère une maladie grave. L’ICGA peut détecter l’inflammation choroïdienne subclinique à des stades très précoces ou même après un traitement systémique.
- Ponction lombaire: La pléocytose dans le liquide céphalo-rachidien est présente chez la majorité des patients atteints de VKH. La ponction lombaire doit être pratiquée tôt dans l’évolution de la maladie, car la pléocytose peut résoudre
Traitement / prise en charge / Lignes directrices
Les objectifs du traitement dans VKH comprennent le diagnostic précoce et la suppression de l’inflammation active, ainsi que la prévention de l’inflammation récurrente et des complications menaçant la vision, telles que le glaucome, le décollement bulleux de la rétine et la néovascularisation choroïdienne.
Le traitement par corticostéroïdes systémiques est le traitement préféré de la maladie VKH, en particulier au stade uvéitique aigu. Il a été démontré que la voie d’administration de corticostéroïdes (orale versus intraveineuse) n’a pas d’impact sur l’acuité visuelle ou l’apparition de complications visuellement significatives dans le traitement de l’HVK aiguë. Pour les maladies graves, le protocole suggéré est l’administration intraveineuse de méthylprednisolone pendant trois jours, suivie d’un traitement oral à forte dose de prednisone. Dans une maladie légère à modérée, la prednisone orale à forte dose peut être suffisante à raison de 1 à 2 mg / kg / jour. La dose de stéroïdes doit être réduite lentement sur environ six mois pour éviter les récidives. Un traitement précoce agressif, associé à des tests FA en série montrant la disparition des fuites de colorant à travers l’EPR, peut aider à prévenir la progression de la maladie, la récurrence et les manifestations extraoculaires. Les stéroïdes topiques et les cycloplégiques peuvent diminuer les cellules de la chambre antérieure et l’humeur vitrée.
Des injections intravitréennes et sous-tenon de triamcinolone ont été utilisées pour le contrôle à court terme de l’inflammation intraoculaire pendant les phases aiguës ou récurrentes; ces thérapies locales doivent être envisagées en cas de maladie récalcitrante et chez les patients qui tolèrent mal les effets secondaires systémiques défavorables des stéroïdes compte tenu de la conicité prolongée des stéroïdes. Les injections intravitréennes d’anti-VEGF sont parfois utilisées pour contrôler la néovascularisation choroïdienne et en cas de détachements rétiniens séreux fovéaux persistants.
Des agents épargneurs de stéroïdes, y compris des antimétabolites, des inhibiteurs de la calcineurine, des produits biologiques, des inhibiteurs du TNF-alpha ou des agents cytotoxiques, peuvent être utilisés pour traiter l’HVK et doivent être surveillés attentivement, souvent en coordination avec un service de rhumatologie. Il y a eu des discussions en cours concernant l’utilisation d’agents immunosuppresseurs non stéroïdiens comme traitement de première intention de la maladie VKH. Cependant, une étude récente n’a révélé aucune différence de résultats entre le traitement immunomodulateur précoce de première intention (TMI) et le traitement à la prednisone seule. De plus, les traitements immunosuppresseurs et biologiques sont coûteux et nécessitent une évaluation préalable minutieuse ainsi qu’un suivi fréquent avec des analyses de sang pour évaluer les effets secondaires graves.
Au stade chronique-récurrent, des récidives fréquentes peuvent suggérer une résistance au traitement par corticostéroïdes et suggérer la nécessité d’un traitement immunomodulateur économe en stéroïdes. L’agent préféré pour les récidives résistantes aux stéroïdes ou l’intolérance aux stéroïdes est la cyclosporine. L’infliximab, le rituximab, l’adalimumab et l’interféron alpha-2a sont des agents biologiques qui ont également été utilisés pour traiter l’uvéite réfractaire dans la maladie VKH.
Pour traiter l’uvéite antérieure souvent associée à une VKH aiguë, des stéroïdes topiques (par exemple, acétate de prednisolone 1%) et une cyloplégie topique (par exemple, cyclopentolate 1% ou atropine 1%) doivent être prescrits en fonction du degré d’inflammation de la chambre antérieure.
Les complications oculaires sont généralement associées à la maladie VKH. Compte tenu des multiples étapes et de la variété des présentations dans lesquelles un patient peut se présenter avec VKH, le traitement peut être retardé dans de nombreux cas. Dans les formes sévères de VKH et dans les récidives, l’inflammation intraoculaire peut être difficile à contrôler et peut entraîner des dommages structurels. Plus de 50% des patients développent des complications connexes, notamment une cataracte, un glaucome secondaire, des membranes néovasculaires choroïdiennes, une fibrose sous-rétinienne ou une combinaison de ces complications (figure 6).
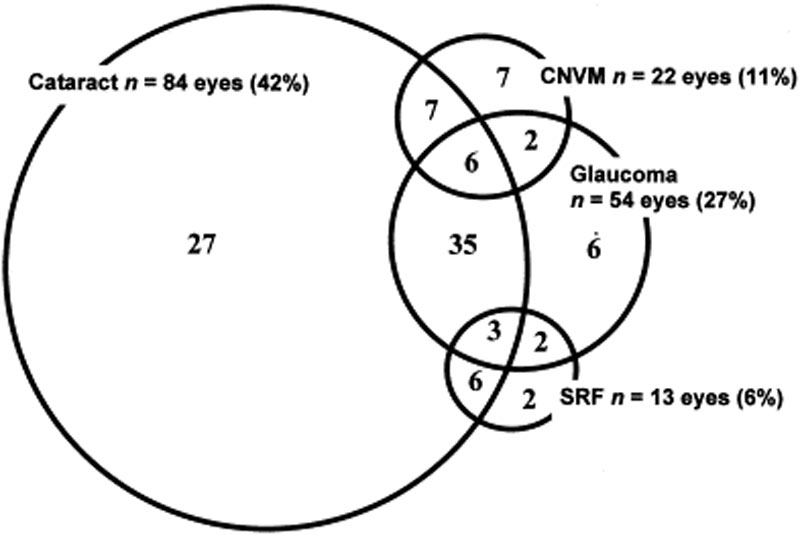
Figure 6: Diagramme de Venn montrant les complications chez les patients atteints de VKH. (Utilisé avec la permission d’Am J Ophthalmol. 2001;131(5):599-606 )
ÉPIDÉMIOLOGIE ET ÉTIOLOGIE
|
SIGNS
|
SYMPTÔMES
|
TRAITEMENT / PRISE EN CHARGE
|
- Du L, Kijlstra A, Yang P. Maladie de Vogt-Koyanagi-Harada: Nouvelles connaissances en physiopathologie, diagnostic et traitement. Prog Retin Eye Res 2016; 52:84-111. https://PubMed.gov/26875727. DOI: 10.1016 / j. preteyeres.2016.02.002
- Les protéines de la famille des Tyrosinases Yamaki K, Gocho K, Hayakawa K, Kondo I, Sakuragi S. sont des antigènes spécifiques de la maladie de Vogt-Koyanagi-Harada. J Immunol 2000; 165 (12): 7323-7329. https://PubMed.gov/11120868
- Horie Y, Takemoto Y, Miyazaki A, Namba K, Kase S, Yoshida K, Ota M, Hasumi Y, Inoko H, Mizuki N, Ohno S. Famille de gènes de la Tyrosinase et maladie de Vogt-Koyanagi-Harada chez les patients japonais. Mol Vis 2006; 12:1601-1605. https://PubMed.gov/17200659
- Ng JY, Luk FO, Lai TY, Pang CP. Influence de la génétique moléculaire dans la maladie de Vogt-Koyanagi-Harada. J Inflamm ophtalmique Infect 2014; 4:20. https://PubMed.gov/25097674. DOI: 10.1186/s12348-014-0020-1
- Bowling B. Uveitis. Kanski’s Clinical Ophthalmology New York, New York: Elsevier; 2016; chapitre 11; p. 395-465.
- Yeh PT YC, Yang CH, Lin CP. Décollement De La Rétine Non segmatogène. Dans: Il s’agit de la première édition de la série télévisée américaine. La rétine de Ryan. Il s’agit de la première édition de la série.
- Goto H RK, Rao N. Maladie de Vogt–Koyanagi–Harada. Dans: Schachat AP SS, Hinton DR, Wilkinson CP, Wiedemann P, éditeur. La rétine de Ryan. Il s’agit de la première édition de la série.
- Riddington L, Hall AJ, Tait B, Nicholson I, Varney M. Syndrome de Vogt-Koyanagi-Harada chez les patients d’ascendance vietnamienne. Aust N Z J Ophthalmol 1996; 24 (2): 147-149. https://PubMed.gov/9199747
- Sugita S, Takase H, Kawaguchi T, Taguchi C, Mochizuki M. Réaction croisée entre les peptides de tyrosinase et l’antigène du cytomégalovirus par les cellules T de patients atteints de la maladie de Vogt-Koyanagi-Harada. Int Ophthalmol 2007; 27 (2-3): 87-95. https://PubMed.gov/17253112. DOI: 10.1007/s10792-006-9020- y
- Freund BK SD, Mieler WF, Yannuzzi LA. Inflammation. L’Atlas Rétinien. Il s’agit de la première édition de la série.
- Maladie de Rao N. Vogt-Koyanagi-Harada. Dans : J YMaD, éditeur. Ophtalmologie. Il s’agit de la première édition de la série télévisée américaine.
- Rao NA, Xu S, Font RL. Ophtalmie sympathique. Une étude immunohistochimique des cellules épithélioïdes et géantes. Ophtalmologie 1985; 92 (12): 1660-1662. https://PubMed.gov/4088616
- Nussenblatt RB. Syndrome de Vogt-Koyanagi-Harada. Dans: Whitcup RBNaSM, éditeur. Uvéite: Principes fondamentaux et Pratique clinique. 4e édition ed: Elsevier; 2010; chapitre Chapitre 24.
- Lire RW, Holland GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L, Pivetti-Pezzi P, Tessler HH, Usui M. Critères diagnostiques révisés pour la maladie de Vogt-Koyanagi-Harada: rapport d’un comité international de nomenclature. Am J Ophthalmol 2001; 131 (5): 647-652. https://PubMed.gov/11336942
- Chung H, Choi DG. Analyse clinique de l’uvéite. Korean J Ophthalmol 1989; 3(1): 33-37. https://PubMed.gov/2795939. DOI: 10.3341/kjo.1989.3.1.33
- Abu El-Asrar AM, Al-Kharashi AS, Aldibhi H, Al-Fraykh H, Kangave D. Maladie de Vogt-Koyanagi-Harada chez les enfants. Oeil (Lond) 2008;22(9):1124-1131. https://PubMed.gov/17479116. DOI: 10.1038/sj.oeil.6702859
- Martin TD, Rathinam SR, Cunningham ET. Prévalence, caractéristiques cliniques et causes de la perte de vision chez les enfants atteints de la maladie de Vogt-Koyanagi-Harada en Inde du Sud. Rétine 2010; 30 (7): 1113-1121. https://PubMed.gov/20168275. DOI: 10.1097/IAE.0b013e3181c96a87
- Forster DJ, Vert RL, Rao NA. Manifestation unilatérale du syndrome de Vogt-Koyanagi-Harada chez un enfant de 7 ans. Am J Ophthalmol 1991; 111(3): 380-382. https://PubMed.gov/2000916
- Maladie de Yamamoto Y, Fukushima A, Nishino K, Koura Y, Komatsu T, Ueno H. Vogt-koyanagi-harada avec apparition chez des patients âgés de 68 à 89 ans. Jpn J Ophthalmol 2007; 51(1): 60-63. https://PubMed.gov/17295144. DOI: 10.1007/s10384-006-0379-0
- Wang Y, Chan CC. Différences entre les sexes dans la maladie de vogt-koyanagi-harada et l’ophtalmie sympathique. J Ophthalmol 2014; 2014: 157803. https://PubMed.gov/24734166. DOI : 10.1155/2014/157803
- Nakao K, Abematsu N, Mizushima Y, Sakamoto T. Gonflement du disque optique dans la maladie de Vogt-Koyanagi-Harada. Invest Ophthalmol Vis Sci 2012; 53 (4): 1917-1922. https://PubMed.gov/22408010. DOI: 10.1167/iovs.11-8984
- Rao NA, Gupta A, Dustin L, Chee SP, Okada AA, Khairallah M, Bodaghi B, Lehoang P, Accorinti M, Mochizuki M, Prabriputaloong T, Lire RW. Fréquence des caractéristiques cliniques distinctives dans la maladie de Vogt-Koyanagi-Harada. Ophthalmology 2010;117(3):591-599, 599.e591. https://PubMed.gov/20036008. DOI: 10.1016/j.ophtha.2009.08.030
- Veerappan M, Fleischman D, Ulrich JN, Stinnett SS, Jaffe GJ, Allingham RR. The Relationship of Vogt-Koyanagi-Harada Syndrome to Ocular Hypertension and Glaucoma. Ocul Immunol Inflamm 2017;25(6):748-752. https://PubMed.gov/27438521. DOI: 10.1080/09273948.2016.1189578
- Baltmr A, Lightman S, Tomkins-Netzer O. Vogt-Koyanagi-Harada syndrome – current perspectives. Clin Ophthalmol 2016;10:2345-2361. https://PubMed.gov/27932857. DOI: 10.2147/OPTH.S94866
- Kitaichi N, Matoba H, Ohno S. Le rôle positif de la ponction lombaire dans le diagnostic de la maladie de Vogt-Koyanagi-Harada: sous-ensembles lymphocytaires dans l’humeur aqueuse et le liquide céphalo-rachidien. Int Ophthalmol 2007; 27 (2-3): 97-103. https://PubMed.gov/17211585. DOI: 10.1007/s10792-006-9016-7
- Oshima Y, Harino S, Hara Y, Tano Y. Résultats angiographiques au vert d’indocyanine dans la maladie de Vogt-Koyanagi-Harada. Am J Ophthalmol 1996; 122(1): 58-66. https://PubMed.gov/8659599
- Lire RW, Yu F, Accorinti M, Bodaghi B, Chee SP, Fardeau C, Goto H, Holland GN, Kawashima H, Kojima E, Lehoang P, Lemaitre C, Okada AA, Pivetti-Pezzi P, Secchi A, Voir RF, Tabbara KF, Usui M, Rao NA. Évaluation de l’effet sur les résultats de la voie d’administration des corticostéroïdes dans la maladie aiguë de Vogt-Koyanagi-Harada. Am J Ophthalmol 2006; 142 (1): 119-124. https://PubMed.gov/16815259. DOI: 10.1016/ j.ajo.2006.02.049
- Rubsamen PE, Gass JD. Syndrome de Vogt-Koyanagi-Harada. Évolution clinique, thérapie et résultat visuel à long terme. Arch Ophthalmol 1991;109(5):682-687. https://PubMed.gov/2025171
- Urzua CA, Velasquez V, Sabat P, Berger O, Ramirez S, Goecke A, Vásquez DH, Gatica H, Guerrero J. Un traitement immunomodulateur antérieur est associé à de meilleurs résultats visuels chez un sous-ensemble de patients atteints de la maladie de Vogt-Koyanagi-Harada. Acta Ophthalmol 2015; 93 (6): e475-480. https://PubMed.gov/25565265. DOI: 10.1111/aos.12648
- Lire RW, Rechodouni A, Butani N, Johnston R, LaBree LD, Smith RE, Rao NA. Complications et facteurs pronostiques dans la maladie de Vogt-Koyanagi-Harada. Am J Ophthalmol 2001; 131 (5): 599-606. https://PubMed.gov/11336934
Format de citation suggéré
Mai AP, Tran C, Wilson CW, Fox AR, Boldt HC. Maladie de Vogt-Koyanagi-Harada (VKH). EyeRounds.org . 1er avril 2019. Disponible à partir de http://EyeRounds.org/cases/284-vogt-koyanagi-harada.htm
Leave a Reply