Enfermedad de Vogt-Koyanagi-Harada (VKH)
Autores: Anthony P. Mai, BS; Charlene Tran, BS; Caroline W. Wilson, MD; Austin R. Fox, MD; H. Culver Boldt, MD
1 de abril de 2019
PRESENTACIÓN INICIAL
Queja principal
Visión borrosa y dolores de cabeza
Antecedentes de Enfermedad Presente
Una mujer vietnamita de 44 años de edad se presentó en el departamento de emergencias con un historial de 10 días de visión borrosa progresiva en ambos ojos y un historial de tres días de dolores de cabeza severos. Su pérdida de visión central no había mejorado con una refracción de su optometrista. Sus fuertes dolores de cabeza occipitales empeoraron con el movimiento y se asociaron con malestar generalizado, fatiga extrema, fotofobia leve y lagrimeo. El paracetamol alivió parcialmente el dolor.
Había viajado recientemente a Vietnam, pero negó haber encontrado contactos enfermos allí. Negó la claudicación de la mandíbula, las fiebres o los cambios de peso. Negó erupciones en la piel, cambios en la audición, tinnitus, mareos, entumecimiento u hormigueo. Negó haber tenido tuberculosis. No tenía antecedentes de problemas de visión previos, afecciones autoinmunes o cáncer.
Antecedentes oculares anteriores
- Antecedentes de cirugía cosmética de párpados (blefaroplastia bilateral) tres años antes
- Sin antecedentes de traumatismo o enfermedad ocular
Antecedentes médicos anteriores
Ninguno
Medicamentos
Paracetamol según sea necesario
Alergias
Sin alergias conocidas a medicamentos
Antecedentes familiares
Sin antecedentes de enfermedad ocular o enfermedad autoinmune
Antecedentes sociales
Emigró de Vietnam varios años antes de la presentación. Está casada y tiene tres hijos. Trabaja en un salón de manicura. No consume productos de tabaco, alcohol ni sustancias ilícitas. Viaja a Vietnam cada seis a doce meses.
Revisión de Sistemas
Negativo, excepto lo que se detalla en la historia de la enfermedad presente
EXAMEN OCULAR
Agudeza visual con/sin corrección (Snellen)
- Ojo derecho (DO): 20/300 (sin mejoría con estenopeico)
- Ojo izquierdo (OS): 20/60-2+2 (sin mejoría con estenopeica)
Motilidad/Alineación ocular
Movimientos extraoculares completos en ambos ojos (OU)
Presión intraocular (PIO): (Tonopen)
- DO: 12 mmHg
- OS: 14 mmHg
Pupilas
- DO: 4 mm en la oscuridad, 3 mm en la luz, sin defecto pupilar aferente relativo (RAPD)
- OS: 4 mm en la oscuridad, 3 mm en la luz, sin RAPD
dedos)
- DO: escotoma central
- OS: Defecto inferotemporal total
Externo
Normal en ambos lados
Examen con lámpara de hendidura
- Tapas/pestañas: Normal
- Conjuntiva/esclerótica: Clara y silenciosa
- Córnea: 1+ epiteliales puntiformes erosiones, no keratic precipita OU
- cámara Anterior: Seguimiento de células y flare y profundo OU
- Iris: arquitectura Normal OU
- Lente: Claro OU
examen con pupila Dilatada (DFE)
- Vítreo: Traza vítreo anterior células OU
- Disco:
- OD: Grado 3 disco edema, hiperemia
- OS: Grado 2-3 disco edema, hiperemia
- Copa-a-disco relación: 0.0 OU
- Mácula:
- OD: 3+ edema macular quístico (EMQ) y líquido subretiniano (SRF) que se extiende desde el disco a la temporal mácula. Sin lípidos ni exudados. Coroides de aspecto pantanoso.
- OS: 2 + CME y SRF extendiéndose desde el disco a través de la fóvea. 1-2 + lípido lineal que se extiende desde el disco hacia la fóvea. Coroides de aspecto pantanoso.
- Vasos:
- OD: Revestimiento temporal
- OS: Normal
- Periferia:
- OD: Quística de la retina mechón anterior del ecuador, a las 10:30
- OS: Superficial SRF anterior del ecuador, a 4:00
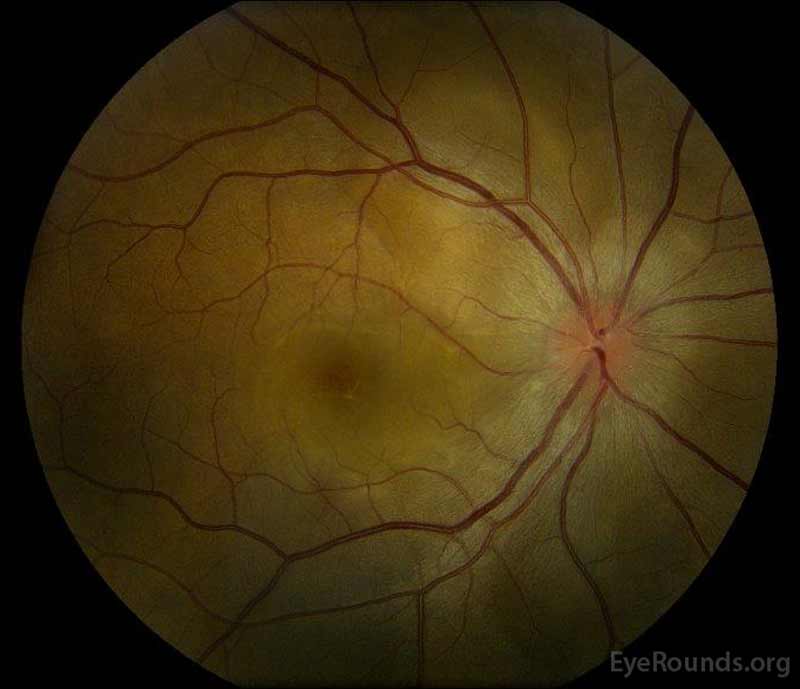 |
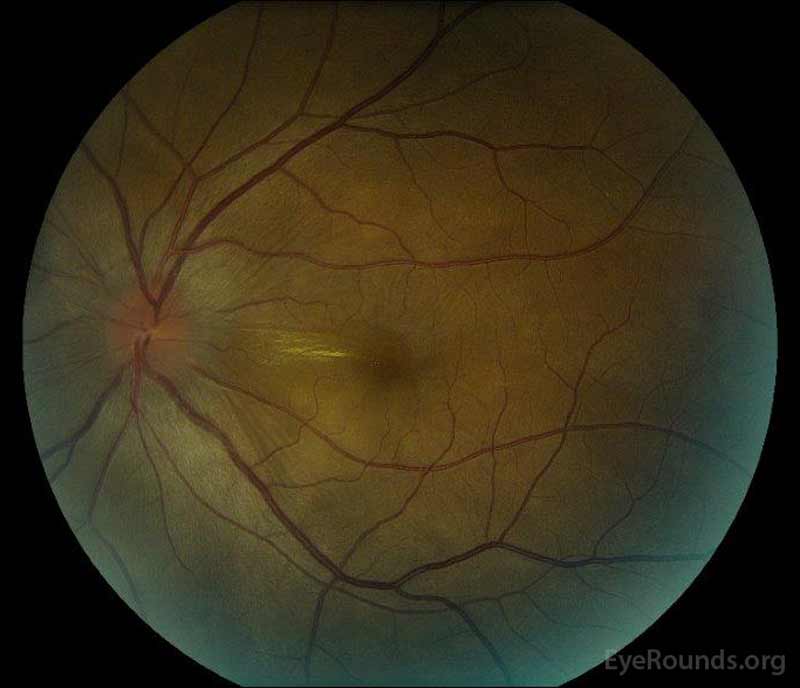 |
Figura 1: Fotografías de fondo de ojo a color en la presentación: (Imagen izquierda) El ojo derecho tiene edema discal e hiperemia leve, así como líquido subretiniano que se extiende desde el disco temporalmente a través de la mácula. También hay un desprendimiento de retina seroso focal superotemporal al disco, a lo largo de la arcada superior. (Right image) The left eye has disc edema and mild hyperemia, along with subretinal fluid extending from the disc to the macula and linear lipid deposits in the nasal macula.
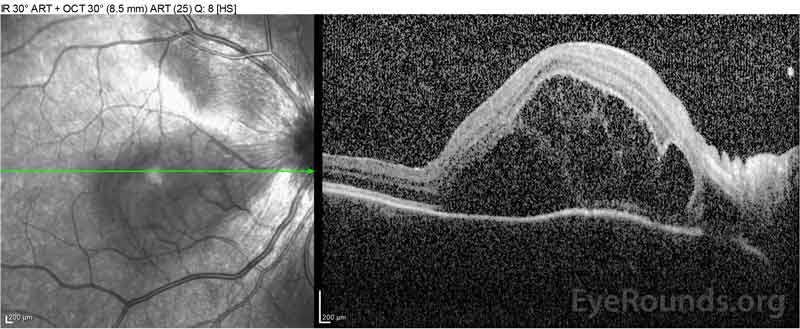 |
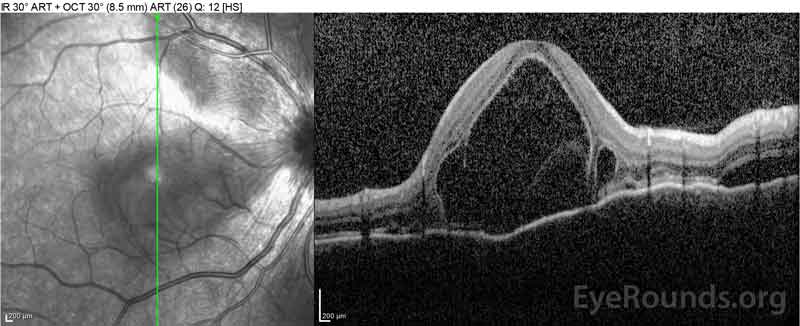 |
 |
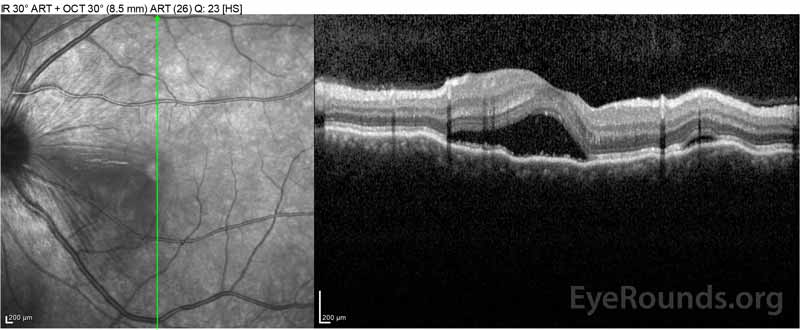 |
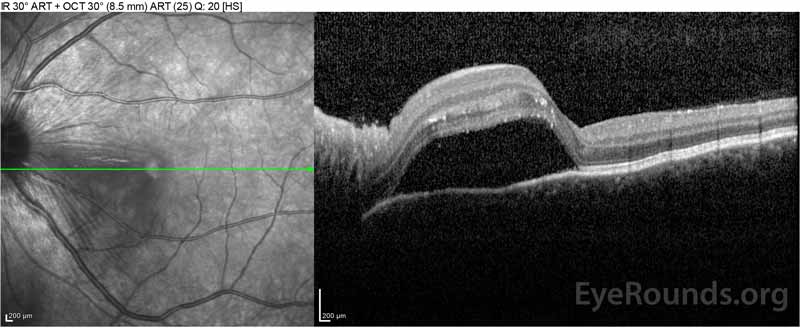
Figure 2: La tomografía de coherencia óptica (OCT) del ojo derecho (paneles superiores) muestra un desprendimiento seroso de retina que involucra la fóvea con líquido intrarretiniano superpuesto extenso, interrupción de las capas externas de la retina y ondulaciones de la coroides engrosada. La OCT del ojo izquierdo (paneles inferiores) muestra un desprendimiento seroso de retina en la mácula nasal que se extiende hasta la fóvea.
 |
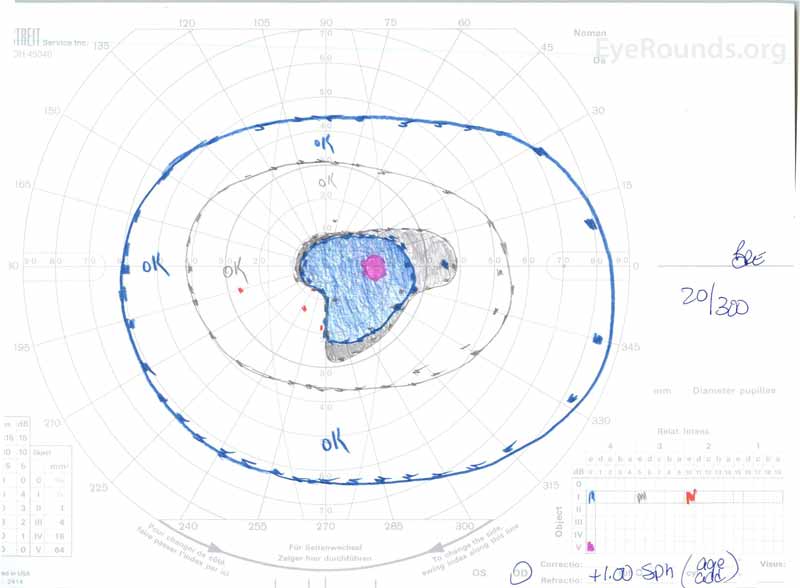 |
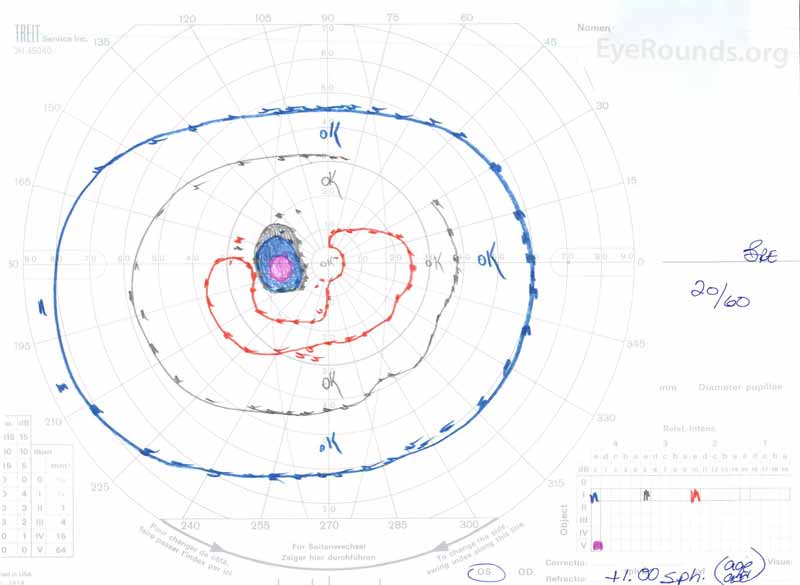
Figura 3: Campos visuales de Goldman (GVF), OU. (Imagen izquierda) La SG muestra un punto ciego fisiológico agrandado y un escotoma central leve. (Imagen derecha) La DO muestra un escotoma central moderadamente denso que incorpora el punto ciego fisiológico y se extiende inferotemporalmente.
B-scan: Sin signos de escleritis, opacidades vítreas leves/células inferiores
Diagnóstico diferencial
- Epiteliopatía pigmentaria placoide multifocal posterior aguda (APMP)
- coriorretinopatía serosa central
- Neuritis óptica
- Panuveitis
- Enfermedad autoinmune (por ejemplo, LES, sarcoidosis)
- Infección (e.ej., sífilis, tuberculosis, Bartonella henselae)
- Malignidad (por ejemplo, linfoma ocular)
- Escleritis posterior
- Oftalmía simpática
- Síndrome de derrame uveal
- Síndrome de Vogt-Koyanagi-Harada
/ p >
Recuento de glóbulos blancos: 4,9 K / mm3 (Ref: 3,7-10,5)
Recuento de glóbulos rojos 3,99 M / mm3 (Ref: 4,0-5,2)
Hemoglobina 11,6 g / dL (Ref: 11,9-15,5)
Hematocrito 35% (Ref: : 35-47)
Recuento de glóbulos blancos: 4,9 K / mm3 (Ref: 3,7-10,5)
Recuento de glóbulos rojos 3,99 M / mm3 (Ref: 4,0-5,2)
Hemoglobina 11,6 g / dL (Ref: 11,9-15,5)
Hematocrito 35% (Ref: : 35-47)
Basic metabolic panel
Sodium 138 mEq/L (Ref: 135-145)
Potassium 4.3 mEq/L (Ref: 3.5-5.0)
Chloride 107 mEq/L (Ref: 95-107)
CO2 20 mEq/L (Ref: 22-29)
Blood urea nitrogen 16 mEq/dL (Ref: 10-20)
Creatinine 0.7 mg/dL (Ref: 0.5-1.0)
C-reactive Protein (CRP): <0.5 mg/dL (Ref: <=0.5)
Erythrocyte sedimentation rate (ESR): 12 mm/Hr (Ref: 0-20)
Angiotensin–converting enzyme (ACE): 13 U/L (Ref: 8-52)
QuantiFERON-TB Oro: Hierro negativo, sangre 54 microgramos / dL (Ref: 37-145)
Capacidad total de fijación de hierro 379 microgramos/dL (Ref: 250-425)
CURSO CLÍNICO
La paciente fue evaluada inicialmente por el servicio de urgencias debido a sus quejas de dolores de cabeza graves de nueva aparición y pérdida de visión. La tomografía computarizada (TC) cerebral y las imágenes por resonancia magnética (RM) no fueron nada notables. La VSG y la PCR estuvieron dentro de los niveles normales. La clínica de oftalmología la evaluó al día siguiente y encontró desprendimientos de retina serosos bilaterales y panuveitis. Los análisis de oro de ACE y QuantiFERON-TB dieron negativo. Se le diagnosticó la enfermedad de Vogt-Koyanagi-Harada en base a su presentación clínica y ascendencia asiática. Recibió tratamiento diario con 80 mg de prednisona, paracetamol según fuera necesario para los dolores de cabeza, y suplementos de vitamina D y calcio. Sus dolores de cabeza se resolvieron rápidamente, y su agudeza visual mejoró constantemente durante las dos semanas siguientes. Su dosis de prednisona se redujo a 40 mg durante tres semanas, con la resolución continua de los síntomas y la mejora de la agudeza visual. No tuvo recurrencia de dolores de cabeza ni empeoramiento de la visión durante la reducción de prednisona. En su cita más reciente, había disminuido a 5 mg cada dos días, sin que reaparecieran los síntomas. Su agudeza visual en esa visita de seguimiento fue de 20/15-2 DO y 20/20+2 SG, y la OCT macular mostró resolución completa de edema discal y desprendimientos serosos de retina en ambos ojos (Figura 4).
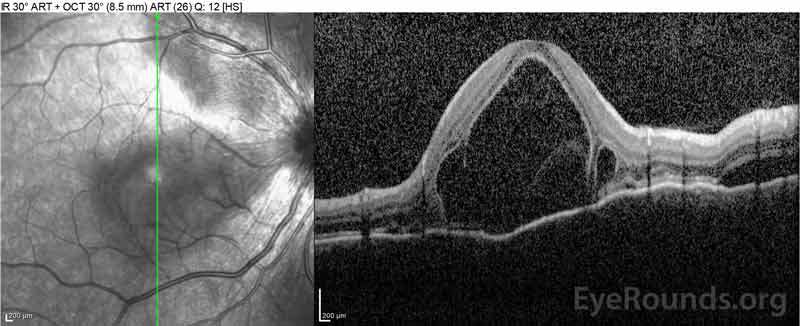
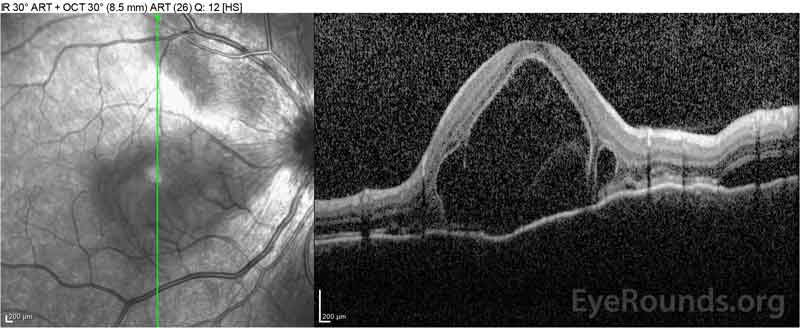
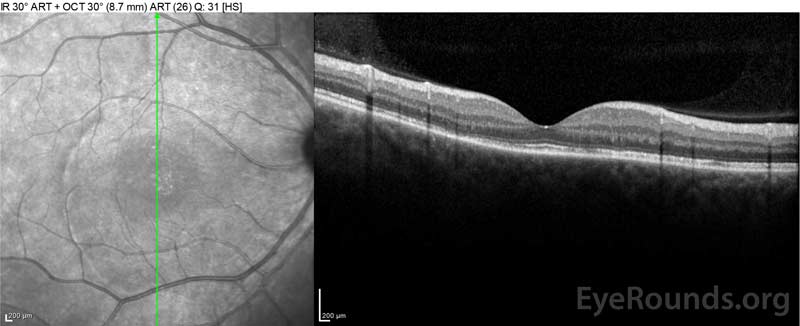
la Figura 4: Tomografía de coherencia óptica que muestra el líquido subretiniano al inicio (parte superior) y el curso de resolución a una semana (media) y cinco semanas (parte inferior) mientras está en una dosis alta de reducción gradual de prednisona oral. Observe el alisado de las ondulaciones coroidales con tratamiento.
 |
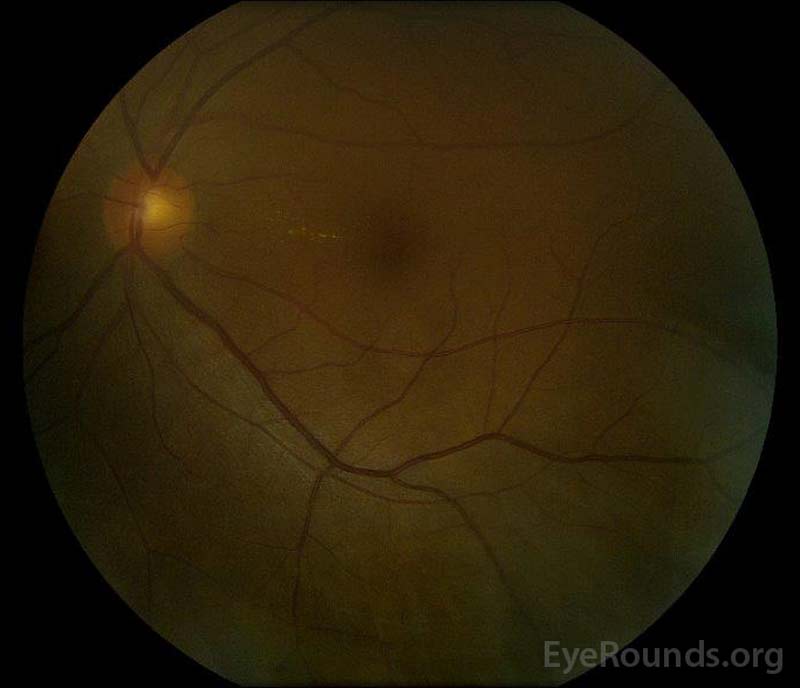 |
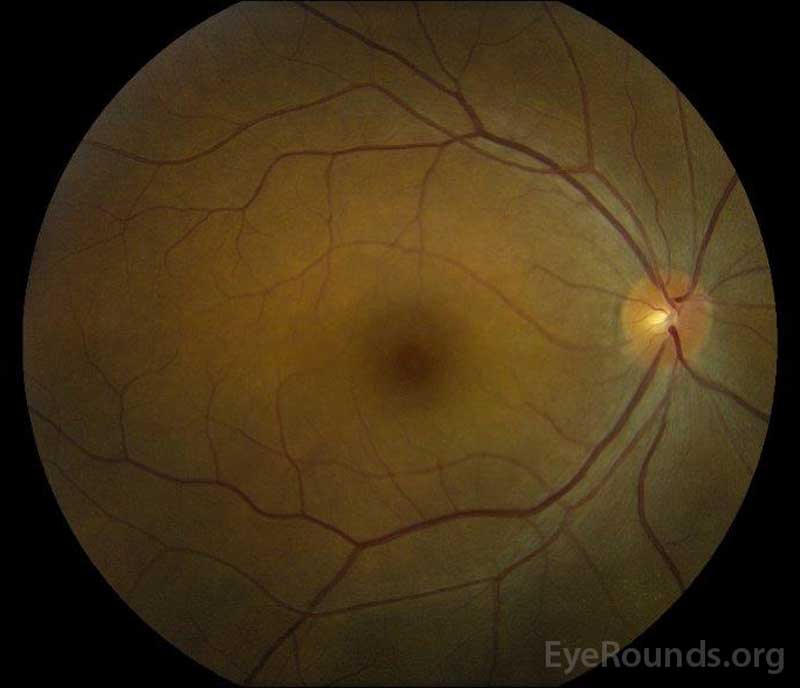
Figura 5: Fotografía del fondo de ojo a color de los ojos derecho (A) e izquierdo (B) durante la fase de convalecencia, lo que demuestra una mejora en el líquido subretiniano y el edema discal.
DIAGNÓSTICO
Enfermedad de Vogt-Koyanagi-Harada incompleta
DISCUSIÓN
La enfermedad de Vogt-Koyanagi-Harada (VKH) es una afección autoinmune sistémica caracterizada por panuveitis granulomatosa bilateral no necrosante asociada con cambios integumentarios extraoculares, como poliosis y vitiligo, e inflamación que afecta a la úvea, el oído interno, el cabello y las meninges. La enfermedad de Harada es la uveítis aislada sin los signos o síntomas sistémicos de VKH.
Etiología
La etiología de la enfermedad de VKH sigue siendo en gran medida desconocida a pesar de los esfuerzos de investigación actuales. Se cree que es una enfermedad autoinmune adquirida que involucra hipersensibilidad mediada por células T a los antígenos melanocíticos, con una predisposición genética subyacente y un posible desencadenante microbiano . La tirosinasa y los péptidos relacionados con la tirosinasa son antígenos de melanocitos que se han sugerido como blancos de procesos autoinmunes en VKH . Sin embargo, el aumento del riesgo de enfermedad de VKH no se asoció con la familia de genes de la tirosinasa, según un estudio .
Debido al aumento de la prevalencia entre ciertos grupos étnicos y de género, se cree que existe una predisposición genética en la patogénesis del VKH. Múltiples genes, entre ellos el antígeno leucocitario humano (HLA) y los genes de interleucina (IL), se han asociado con VKH en diferentes poblaciones étnicas . Los receptores HLA son complejos de histocompatibilidad importantes en humanos que presentan péptidos al sistema inmunitario. HLA-DR1, HLA-DR4, HLA-DRB1 * 0405 y HLA-DRw53 son varios haplotipos que se encuentran en pacientes con enfermedad de VKH . El HLA-DR4 es más común en japoneses e hispanos, mientras que el HLA-DRB1*0405 es más frecuente en pacientes coreanos y de Oriente Medio . Los alelos HLA-DR4 y HLA-DRB1 * 0405 se encuentran en pacientes vietnamitas . El receptor HLA-DRB1 se une a los antígenos de melanocitos en diferentes capacidades. A pesar de estas asociaciones, las pruebas genéticas no se recomiendan en este momento.
Dados los síntomas prodrómicos habituales que acompañan al VKH, como fiebre, dolores de cabeza, meningismo y tinnitus, se ha sugerido una etiología viral incitante como desencadenante de la aparición del VKH a través de mecanismos de mimetismo molecular en pacientes genéticamente predispuestos. La glicoproteína H de la envoltura del citomegalovirus tiene una homología significativa de aminoácidos con el péptido de tirosinasa, y la infección por CMV puede desencadenar VKH a través de la imitación molecular (es decir, el reconocimiento por receptores de clase II de HLA) . También se ha implicado el virus Ebstein-bar (VEB). Sin embargo, no ha habido evidencia definitiva con respecto a la etiología viral del VKH, y sigue sin estar claro qué desencadena la respuesta autoinmune del VKH .
Fisiopatología
Hay cuatro fases clásicas de VKH que pueden tener presentaciones variables: prodrómica, uveítica aguda, convaleciente y crónica recurrente. Los cambios histopatológicos suelen comenzar en la fase aguda .
La fase uveítica aguda se caracteriza por engrosamiento uveal bilateral secundario a inflamación granulomatosa. Los granulomas consisten en linfocitos, macrófagos y células epitelioides y gigantes llenas de gránulos . Aunque anteriormente se creía que las células epitelioides eran melanocitos alterados, un estudio inmunohistoquímico de seguimiento sugirió un origen de macrófagos de tejido en su lugar . Los granulomas llenos de histiocitos epitelioides, denominados nódulos de Dalen-Fuchs, a menudo se pueden ver entre el epitelio pigmentario de la retina (EPR) y la membrana de Bruch. La inflamación granulomatosa uveal conduce al engrosamiento coroideo y desprendimientos exudativos de retina llenos de líquido proteico. Además, la pleocitosis (i.e., aumento del recuento de células) puede estar presente en la cámara anterior y en el vítreo .
La fase de convalecencia se identifica por la despigmentación de las áreas coroides y extraoculares, incluida la piel y el cabello perioculares. Una coroides despigmentada contra un nervio óptico pálido da la impresión de un fondo de ojo» que brilla al atardecer», que es una característica clásica de esta fase del VKH . Además, los nódulos de Dalen-Fuchs se vuelven más prominentes debajo de la EPR en la fase de convalecencia .
La fase crónica recurrente se caracteriza por una disminución del grosor coroidal, resolución de desprendimientos serosos de retina, vitritis crónica leve e inflamación recurrente del segmento anterior granulomatoso. La neovascularización coroidea (NVC) y la fibrosis subretiniana pueden desarrollarse durante esta fase y son indicadores de progresión grave de la enfermedad . Las cataratas y el glaucoma secundario son otras complicaciones de la inflamación recurrente o de larga duración en esta fase .
Epidemiología
El VKH es frecuente en razas con pigmento de piel más oscura, especialmente Asiáticos, Sudamericanos, del Medio Oriente y nativos americanos. La enfermedad de VKH representa > el 10% de la uveítis en estas poblaciones . Se cree que solo del 1 al 4% de los casos de uveítis son secundarios a la enfermedad de VKH en los Estados Unidos (7). En los Estados Unidos, se ha encontrado que la mayoría de los casos de VKH afectan a personas decentes de origen asiático, hispano o nativo americano . Curiosamente, la enfermedad de VKH rara vez afecta a los africanos a pesar de su pigmentación oscura . La incidencia de la enfermedad de VKH varía mucho entre los subgrupos raciales de los países vecinos . Por ejemplo, la incidencia de VKH en Corea es de solo el 2%, muy inferior a la observada en Japón y China .
El VKH tiene un inicio típico de 20 a 50 años de edad ; sin embargo, los estudios sugieren que 3,1-13,4% de los casos de VKH son pacientes pediátricos y 10% de los casos son ≥65 años de edad . Clásicamente, se cree que el VKH tiene predilección por el género femenino, y aunque la mayoría de los estudios muestran que el VKH afecta desproporcionadamente a las mujeres, algunos estudios han mostrado una predisposición masculina o ninguna predisposición de género .
Signos / síntomas
Como se mencionó anteriormente, los cuatro estadios de la enfermedad de VKH son prodrómica, uveítica, convaleciente y crónica recurrente. Cada etapa presenta características clínicas distintas.
- Prodrómico: Esta etapa inicial puede presentarse como una enfermedad de tipo gripal con síntomas predominantemente constitucionales, como dolor de cabeza, mareos, fiebre, fatiga y/o náuseas. Se han notificado síntomas neurológicos de meningitis, parálisis de los nervios craneales y neuritis óptica, así como síntomas auditivos de tinnitus, disacusia y vértigo . La fotofobia, la visión borrosa, las moscas volantes y/o el dolor ocular generalmente comienzan dentro de las 48 horas posteriores a los síntomas prodrómicos . La fase prodrómica suele durar de unos pocos días a semanas. Uveítis aguda: Esta etapa incluye visión borrosa, fotofobia, inyección conjuntival y dolor ocular. Puede haber uveítis anterior leve que al principio parece no granulomatosa. El inicio unilateral generalmente cambia a compromiso bilateral en 1-2 semanas. Se puede desarrollar uveítis anterior granulomatosa con precipitados queráticos de grasa de cordero. Los hallazgos del examen posterior pueden incluir edema e hiperemia del nervio óptico, áreas multifocales de coroiditis, múltiples áreas de desprendimientos serosos de retina localizados en el fondo del ojo posterior, engrosamiento coroideo, pliegues coriorretinianos radiantes y vitritis . Los desprendimientos serosos de retina pueden formar un patrón de hojas de trébol en el fondo posterior y pueden progresar a desprendimientos ampollosos extensos en casos graves . El glaucoma inflamatorio agudo se ha asociado con esta fase de la enfermedad y puede presentarse con una cámara anterior superficial secundaria a edema del cuerpo ciliar, imitando el cierre de ángulo agudo . La duración de la fase uveítica aguda depende del diagnóstico y tratamiento oportunos.
- Uveítis crónica o Convaleciente: Esta etapa se desarrolla típicamente varias semanas después de la fase aguda y se caracteriza por vitiligo (por ejemplo, cara, manos, hombros o espalda), poliosis y alopecia . La despigmentación cerca del limbo corneal, conocida como signo de Sugiura, se puede ver un mes después del inicio de la enfermedad ; sin embargo, este signo rara vez se ve fuera de la población japonesa . La despigmentación coroidea generalmente ocurre en unos pocos meses y da como resultado el color rojo anaranjado brillante de la coroides y el fondo clásico «sunset glow fundus».»Se cree que el fondo Sunset glow es el más importante y predictivo en el diagnóstico de VKH crónico . En la periferia media se pueden formar cicatrices coriorretinianas redondeadas, bien definidas y numulares. La fase uveítica crónica suele durar varios meses.
- Crónica recurrente: Esta etapa se caracteriza por episodios recurrentes de uveítis anterior granulomatosa con precipitados queráticos de grasa de cordero, nódulos del iris, despigmentación del iris, sinequias posteriores, cataratas subcapsulares posteriores, glaucoma secundario, membranas neovasculares coroideas y, en última instancia, fibrosis subretiniana y atrofia coriorretiniana numular . La fase crónica generalmente se desarrolla al menos seis meses después de la presentación inicial. Los desprendimientos serosos de retina presentes durante las fases aguda y convaleciente generalmente no se repiten con un tratamiento agresivo con corticosteroides .
Criterios Diagnósticos
Los criterios diagnósticos más recientes, denominados Criterios Diagnósticos Revisados (CDR) para VKH, se definieron en 1999 en el Primer Taller Internacional sobre VKH . Estos se describen en el cuadro 1. Los RDC son útiles en el sentido de que dividen el VKH en tres categorías diagnósticas diferentes basadas en la fase de la enfermedad durante la cual el paciente se presenta: completo, incompleto y probable. Esta categorización de la enfermedad permite un manejo apropiado y temprano de la enfermedad » probable «que puede ayudar a prevenir la progresión a la enfermedad» completa».
Los análisis para otras causas de inflamación ocular, tanto infecciosas como autoinflamatorias, son esenciales. Estos pueden incluir velocidad de sedimentación de eritrocitos( VSG), proteína C reactiva (PCR), pruebas de cuantiferon-Gold para tuberculosis, reactiva plasmática rápida (RPR) para sífilis, enzima convertidora de angiotensina (ECA) y una radiografía de tórax para sarcoidosis, anticuerpo antinuclear (ANA) y ANCA p/c. Además, se debe anotar un historial de traumatismo ocular o cirugía intraocular reciente y probablemente sugiera oftalmia simpática (SO) como el diagnóstico más probable, dada la presentación y fisiopatología muy similares que comparten SO y VKH .
Para apoyar un diagnóstico de VKH en casos equívocos, se puede realizar una punción lumbar para buscar pleocitosis linfocítica y monocítica; sin embargo, esto rara vez se emplea clínicamente. El ochenta por ciento de los pacientes tiene pleocitosis en el líquido cefalorraquídeo (LCR) en una semana y el 97% tiene pleocitosis en tres semanas. El aumento de los niveles de células inmunitarias puede durar hasta ocho semanas después de la aparición de la enfermedad . Los perfiles de marcadores de superficie de células T son similares entre el LCR y el humor acuoso, pero diferentes de la sangre. Esto sugiere la capacidad del LCR para reflejar con precisión la inflamación uveal en la enfermedad de VKH .
Cuadro 1. Criterios Diagnósticos Revisados para Enfermedad de Vogt-Koyanagi-Harada
*De la Tabla 1 en (15).
«Enfermedad completa de Vogt-Koyanagi-Harada (los criterios 1 a 5 deben estar presentes)
- Sin antecedentes de traumatismo ocular penetrante o cirugía antes de la aparición inicial de uveítis.
- No hay evidencia clínica o de laboratorio que sugiera otras entidades de enfermedad ocular.
- Compromiso ocular bilateral (se debe cumplir a o b, dependiendo del estadio de la enfermedad en el que se examine al paciente).
- Manifestaciones tempranas de la enfermedad.
- Debe haber evidencia de una coroiditis difusa (con o sin uveítis anterior, reacción inflamatoria vítrea o hiperemia del disco óptico), que puede manifestarse como una de las siguientes:
- Manifestaciones tempranas de la enfermedad.
- Áreas focales de líquido subretiniano, o
- Desprendimientos de retina serosos bullosos.
- Con hallazgos equívocos del fondo de ojo; también deben estar presentes los dos elementos siguientes:
- Áreas focales de retraso en la perfusión coroidal, áreas multifocales de fuga puntual, áreas placoides grandes de hiperfluorescencia, acumulación dentro del líquido subretiniano y tinción del nervio óptico (enumeradas en orden de aparición secuencial) mediante angiografía con fluoresceína, y
- Engrosamiento coroidal difuso, sin evidencia de escleritis posterior mediante ecografía.
- Manifestaciones tardías de la enfermedad.
- Antecedentes que sugieren la presencia previa de hallazgos de 3a, y ambos (2) y (3) a continuación o múltiples signos de (3):
- Despigmentación ocular (cualquiera de las siguientes manifestaciones es suficiente): (a) Fondo de resplandor de la puesta de sol, o (b) signo de Sugiura.
- Otros signos oculares:
- Cicatrices despigmentadas coriorretinianas numulares, o
- Aglomeración y/o migración del epitelio pigmentario retiniano, o
- Uveítis anterior recurrente o crónica.
- Hallazgos neurológicos / auditivos(pueden haberse resuelto en el momento del examen).
- Meningismo (malestar, fiebre, dolor de cabeza, náuseas, dolor abdominal, rigidez del cuello y la espalda, o una combinación de estos factores; sin embargo, el dolor de cabeza por sí solo no es suficiente para cumplir con la definición de meningismo), o
- Tinnitus, o
- pleocitosis de líquido cefalorraquídeo.
- Hallazgo integumentario (que no precede al inicio de la enfermedad ocular o del sistema nervioso central).
- Alopecia, o
- Poliosis, o
- Vitiligo.
Enfermedad de Vogt-Koyanagi-Harada incompleta (los criterios 1 a 3 y 4 o 5 deben estar presentes)
- No hay antecedentes de traumatismo ocular penetrante o cirugía antes del inicio inicial de la uveítis, y
- No hay evidencia clínica o de laboratorio que sugiera otras entidades de enfermedad ocular, y
- Afectación ocular bilateral.
- Hallazgos neurológicos / auditivos; como se define para la enfermedad completa de Vogt-Koyanagi-Harada arriba, o
- Hallazgos integumentarios; como se define para la enfermedad completa de Vogt-Koyanagi-Harada arriba.
Probable enfermedad de Vogt-Koyanagi-Harada (enfermedad ocular aislada; los criterios 1 a 3 deben estar presentes)
- Sin antecedentes de traumatismo ocular penetrante o cirugía antes de la aparición inicial de uveítis.
- No hay evidencia clínica o de laboratorio que sugiera otras entidades de enfermedad ocular.
- Compromiso ocular bilateral, tal como se ha definido anteriormente para la enfermedad completa de Vogt-Koyanagi-Harada. «
Pruebas / Análisis de laboratorio
En el análisis inicial de VKH, se debe considerar la obtención de las siguientes pruebas:
- Tomografía de coherencia óptica (OCT): En la fase uveítica aguda, la OCT probablemente mostrará un engrosamiento coroideo significativo y desprendimientos serosos de retina. Las acumulaciones de líquido subretiniano pueden tener septaciones que se cree que son membranas de fibrina y productos inflamatorios, creando una estructura lobular que también se puede ver en la angiografía con fluoresceína. En la fase de convalecencia, la OCT puede detectar áreas de adelgazamiento de la retina después de la inflamación resuelta después del tratamiento con corticosteroides .
- Ecografía B-scan: En la fase aguda, la ecografía puede mostrar engrosamiento coroidal posterior difuso, engrosamiento escleral posterior, desprendimientos de retina y opacidades vítreas . Se pueden observar derrames ciliares con biomicroscopia ecográfica . Esta prueba también es útil para descartar la escleritis posterior.
- Angiografía con fluoresceína (FA): Clásicamente, la FA revela puntos hipofluorescentes coroidales multifocales en la fase temprana, seguidos de múltiples áreas hiperfluorescentes focales con fuga difusa en la fase tardía . El tinte se filtra a través del RPE y se acumula en el espacio subretiniano que rodea los puntos hiperfluorescentes. La AF puede ser útil para el diagnóstico cuando la enfermedad de VKH se presenta sin síntomas extraoculares. Hiperfluorescencia del disco óptico y defectos de ventana causados por cicatrices coriorretinales atróficas se pueden ver en la periferia media . La AF en la etapa crónica recurrente de la enfermedad de VKH muestra defectos de ventana inespecíficos debido al daño de la EPR, la neovascularización coroidea y la fibrosis subretiniana .Angiografía con verde de indocianina (ICG) : El GIC de fase temprana muestra vasos estromales hiperfluorescentes que indican vasculopatía coroidea y puntos oscuros hipofluorescentes que corresponden a granulomas y relleno irregular retardado de la vasculatura coroidea . La fase tardía revela patrones vasculares estromales difusos e hiperfluorescencia coroidea difusa. La hiperfluorescencia del disco sugiere una enfermedad grave. ICGA puede detectar inflamación coroidea subclínica en etapas muy tempranas o incluso después de la terapia sistémica .Punción lumbar: La pleocitosis en el líquido cefalorraquídeo está presente en la mayoría de los pacientes con VKH. La punción lumbar se debe realizar al principio del curso de la enfermedad, ya que la pleocitosis puede resolver
Tratamiento/Manejo/Pautas
Los objetivos del tratamiento en VKH incluyen el diagnóstico precoz y la supresión de la inflamación activa, junto con la prevención de la inflamación recurrente y las complicaciones que ponen en peligro la visión, como glaucoma, desprendimiento de retina ampolloso y neovascularización coroidea.
El tratamiento sistémico con corticosteroides es la terapia preferida para la enfermedad de VKH, especialmente durante el estadio uveítico agudo. Se ha demostrado que la vía de administración de corticosteroides (oral frente a intravenosa) no afecta la agudeza visual ni la aparición de complicaciones visualmente significativas en el tratamiento del VKH agudo . Para la enfermedad grave, el protocolo sugerido es la administración intravenosa de metilprednisolona durante tres días, seguida de un tratamiento oral de dosis altas de prednisona. En la enfermedad leve a moderada, la prednisona oral en dosis altas puede ser suficiente a dosis de 1-2 mg/kg / día. La dosis de esteroides debe reducirse lentamente durante aproximadamente seis meses para evitar la recurrencia . El tratamiento temprano agresivo, junto con pruebas de FA en serie que muestran la desaparición de la fuga de tinte a través de la EPR, puede ayudar a prevenir la progresión de la enfermedad, la recurrencia y las manifestaciones extraoculares . Los esteroides tópicos y los ciclopléjicos pueden disminuir las células de la cámara anterior y el humor vítreo.
Las inyecciones intravítreas y por debajo de la espiga de triamcinolona se han utilizado para el control a corto plazo de la inflamación intraocular durante las fases aguda o recurrente; estas terapias locales se deben considerar en el caso de enfermedad recalcitrante y en pacientes que toleran mal los efectos secundarios sistémicos desfavorables de los esteroides, dada la reducción progresiva de los esteroides. Las inyecciones intravítreas anti-VEGF se utilizan a veces para el control de la neovascularización coroidea y en casos de desprendimientos de retina serosos foveales persistentes .
Los agentes ahorradores de esteroides, incluidos antimetabolitos, inhibidores de calcineurina, productos biológicos, inhibidores de TNF-alfa o agentes citotóxicos, se pueden utilizar para tratar el VKH y deben monitorizarse cuidadosamente, a menudo en coordinación con un servicio de reumatología . Ha habido un debate en curso sobre el uso de agentes inmunosupresores no esteroideos como terapia de primera línea para la enfermedad de VKH. Sin embargo, un estudio reciente no reveló diferencias en los resultados entre el tratamiento inmunomodulador de primera línea (IMT) temprano y el tratamiento con prednisona sola . Además, las terapias inmunosupresoras y biológicas son costosas y requieren una cuidadosa evaluación previa al tratamiento, así como un seguimiento frecuente con análisis de sangre para evaluar los efectos secundarios graves.
En la etapa de recidiva crónica, la recidiva frecuente puede sugerir resistencia a la terapia con corticosteroides y sugiere la necesidad de un tratamiento inmunomodulador ahorrador de esteroides . El agente preferido para la recidiva resistente a los esteroides o la intolerancia a los esteroides es la ciclosporina . Infliximab, rituximab, adalimumab e interferón alfa-2a son fármacos biológicos que también se han utilizado para tratar la uveítis refractaria en la enfermedad de VKH.
Para tratar la uveítis anterior a menudo asociada con VKH aguda, se deben prescribir esteroides tópicos (por ejemplo, acetato de prednisolona al 1%) y cicloplejía tópica (por ejemplo, ciclopentolato al 1% o atropina al 1%) dependiendo del grado de inflamación de la cámara anterior.
Las complicaciones oculares se asocian comúnmente con la enfermedad de VKH. Dadas las múltiples etapas y la variedad de presentaciones en las que un paciente puede presentar VKH, el tratamiento puede retrasarse en muchos casos. En formas graves de VKH y en recidivas, la inflamación intraocular puede ser difícil de controlar y puede resultar en daño estructural. Más del 50% de los pacientes desarrollan complicaciones relacionadas, como cataratas, glaucoma secundario, membranas neovasculares coroideas, fibrosis subretiniana o una combinación de estas (Figura 6) .
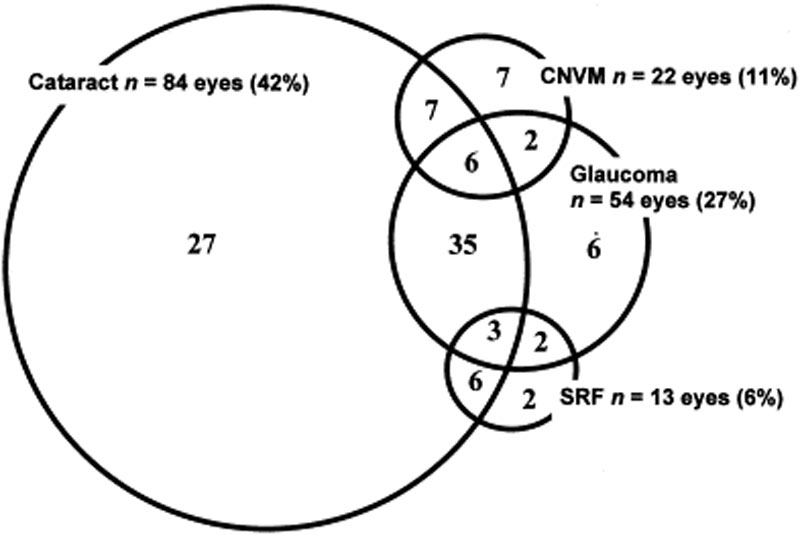
Figura 6: Diagrama de Venn que muestra complicaciones en pacientes con VKH. (Utilizado con permiso de Am J Ophthalmol. 2001;131(5):599-606 )
EPIDEMIOLOGÍA Y ETIOLOGÍA
|
SIGNS
|
SÍNTOMAS
|
TRATAMIENTO/MANEJO
|
- Du L, Kijlstra A, Yang P. Vogt-Koyanagi-Harada disease: Novel insights into pathophysiology, diagnosis and treatment (en inglés). Prog Retin Eye Res 2016; 52: 84-111. https://PubMed.gov/26875727. DOI: 10.1016 / j. preteyeres.2016.02.002
- Las proteínas de la familia de la tirosinasa Yamaki K, Gocho K, Hayakawa K, Kondo I, Sakuragi S. son antígenos específicos de la enfermedad de Vogt-Koyanagi-Harada. J Immunol 2000; 165 (12): 7323-7329. https://PubMed.gov/11120868
- Horie Y, Takemoto Y, Miyazaki A, Namba K, Kase S, Yoshida K, Ota M, Hasumi Y, Inoko H, Mizuki N, Ohno S. Familia de genes de la tirosinasa y enfermedad de Vogt-Koyanagi-Harada en pacientes japoneses. Mol Vis 2006; 12: 1601-1605. https://PubMed.gov/17200659
- Ng JY, Luk FO, Lai TY, Pang CP. Influencia de la genética molecular en la enfermedad de Vogt-Koyanagi-Harada. J Inflamación oftálmica 2014;4:20. https://PubMed.gov/25097674. DOI: 10.1186 / s12348-014-0020-1
- Bowling B. Uveítis. Kanski Clinical Ophthalmology Nueva York, Nueva York: Elsevier; 2016; capítulo 11; p. 395-465.
- Yeh PT YC, Yang CH, Lin CP. Desprendimiento de Retina No Regmatógeno. En: Schachat AP SS, Hinton DR, Wilkinson CP, Wiedemann P,, editor. La Retina de Ryan. Nueva York: Elsevier; 2018; capítulo 99; p. 1828-1849.
- Goto H RK, Rao N. Enfermedad de Vogt–Koyanagi-Harada. En: Schachat AP SS, Hinton DR, Wilkinson CP, Wiedemann P, editor. La Retina de Ryan. Nueva York, Nueva York: Elsevier; 2018; capítulo 78; p. 1505-1515.
- Síndrome de Riddington L, Hall AJ, Tait B, Nicholson I, Varney M. Vogt-Koyanagi-Harada en pacientes de ascendencia vietnamita. Aust N Z J Ophthalmol 1996; 24(2): 147-149. https://PubMed.gov/9199747
- Sugita S, Takase H, Kawaguchi T, Taguchi C, Mochizuki M. Reacción cruzada entre los péptidos de la tirosinasa y el antígeno del citomegalovirus por células T de pacientes con enfermedad de Vogt-Koyanagi-Harada. Int Ophthalmol 2007;27 (2-3): 87-95. https://PubMed.gov/17253112. DOI: 10.1007 / s10792-006-9020-y
- Freund BK SD, Mieler WF, Yannuzzi LA. Inflamación. El Atlas de la Retina. Nueva York, Nueva York: Elsevier 2017; capítulo 4; p. 279-398.
- Enfermedad de Rao N. Vogt-Koyanagi-Harada. En: J YMaD, editor. Oftalmología. Nueva York, Nueva York: Elsevier; 2014; capítulo 7.17; págs. 761 a 763.
- Rao NA, Xu S, Font RL. Oftalmia simpática. Estudio inmunohistoquímico de células epitelioides y gigantes. Ophthalmology 1985; 92 (12): 1660-1662. https://PubMed.gov/4088616
- Nussenblatt RB. Síndrome de Vogt-Koyanagi-Harada. En: Whitcup RBNaSM, editor. Uveítis: Fundamentos y Práctica Clínica. 4a Edición ed: Elsevier; 2010; capítulo Capítulo 24.
- Read RW, Holland GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L, Pivetti-Pezzi P, Tessler HH, Usui M. Criterios diagnósticos revisados para la enfermedad de Vogt-Koyanagi-Harada: informe de un comité internacional de nomenclatura. Am J Ophthalmol 2001; 131 (5): 647-652. https://PubMed.gov/11336942
- Chung H, Choi DG. Análisis clínico de uveítis. Korean J Ophthalmol 1989; 3 (1): 33-37. https://PubMed.gov/2795939. DOI: 10.3341 / kjo.1989.3.1.33
- Enfermedad de Abu El-Asrar AM, Al-Kharashi AS, Aldibhi H, Al-Fraykh H, Kangave D. Vogt-Koyanagi-Harada en niños. Ojo (Lond) 2008;22(9):1124-1131. https://PubMed.gov/17479116. DOI: 10.1038 / sj.ojo.6702859
- Martin TD, Rathinam SR, Cunningham ET. Prevalence, clinical characteristics, and causes of vision loss in children with Vogt-Koyanagi-Harada disease in South India. Retina 2010;30 (7): 1113-1121. https://PubMed.gov/20168275. DOI: 10.1097 / IAE.0b013e3181c96a87
- Forster DJ, Green RL, Rao NA. Manifestación unilateral del síndrome de Vogt-Koyanagi-Harada en un niño de 7 años. Am J Ophthalmol 1991; 111 (3): 380-382. https://PubMed.gov/2000916
- Enfermedad de Yamamoto Y, Fukushima A, Nishino K, Koura Y, Komatsu T, Ueno H. Vogt-koyanagi-harada con inicio en pacientes de edad avanzada de 68 a 89 años. Jpn J Ophthalmol 2007; 51 (1): 60-63. https://PubMed.gov/17295144. DOI: 10.1007 / s10384-006-0379-0
- Wang Y, Chan CC. Diferencias de género en la enfermedad de vogt-koyanagi-harada y la oftalmia simpática. J Ophthalmol 2014; 2014: 157803. https://PubMed.gov/24734166. DOI: 10.1155/2014/157803
- Nakao K, Abematsu N, Mizushima Y, Sakamoto T. Inflamación del disco óptico en la enfermedad de Vogt-Koyanagi-Harada. Invest Ophthalmol Vis Sci 2012; 53 (4): 1917-1922. https://PubMed.gov/22408010. DOI: 10.1167 / iovs.11-8984
- Rao NA, Gupta A, Dustin L, Chee SP, Okada AA, Khairallah M, Bodaghi B, Lehoang P, Accorinti M, Mochizuki M, Prabriputaloong T, Read RW. Frecuencia de las características clínicas distintivas en la enfermedad de Vogt-Koyanagi-Harada. Ophthalmology 2010;117(3):591-599, 599.e591. https://PubMed.gov/20036008. DOI: 10.1016/j.ophtha.2009.08.030
- Veerappan M, Fleischman D, Ulrich JN, Stinnett SS, Jaffe GJ, Allingham RR. The Relationship of Vogt-Koyanagi-Harada Syndrome to Ocular Hypertension and Glaucoma. Ocul Immunol Inflamm 2017;25(6):748-752. https://PubMed.gov/27438521. DOI: 10.1080/09273948.2016.1189578
- Baltmr A, Lightman S, Tomkins-Netzer O. Vogt-Koyanagi-Harada syndrome – current perspectives. Clin Ophthalmol 2016;10:2345-2361. https://PubMed.gov/27932857. DOI: 10.2147/OPTH.S94866
- Kitaichi N, Matoba H, Ohno S. El papel positivo de la punción lumbar en el diagnóstico de la enfermedad de Vogt-Koyanagi-Harada: subconjuntos de linfocitos en el humor acuoso y el líquido cefalorraquídeo. Int Ophthalmol 2007; 27 (2-3): 97-103. https://PubMed.gov/17211585. DOI: 10.1007 / s10792-006-9016-7
- Oshima Y, Harino S, Hara Y, Tano Y. Hallazgos angiográficos en verde de indocianina en la enfermedad de Vogt-Koyanagi-Harada. Am J Ophthalmol 1996; 122 (1): 58-66. https://PubMed.gov/8659599
- Read RW, Yu F, Accorinti M, Bodaghi B, Chee SP, Fardeau C, Goto H, Holland GN, Kawashima H, Kojima E, Lehoang P, Lemaitre C, Okada AA, Pivetti-Pezzi P, Secchi A, See RF, Tabbara KF, Usui M, Rao NA. Evaluación del efecto sobre los resultados de la vía de administración de corticosteroides en la enfermedad aguda de Vogt-Koyanagi-Harada. Am J Ophthalmol 2006; 142 (1): 119-124. https://PubMed.gov/16815259. DOI: 10.1016 / j. ajo.2006.02.049
- Rubsamen PE, Gass JD. Síndrome de Vogt-Koyanagi-Harada. Evolución clínica, terapia y resultado visual a largo plazo. Arch Ophthalmol 1991;109(5):682-687. https://PubMed.gov/2025171
- Urzua CA, Velasquez V, Sabat P, Berger O, Ramirez S, Goecke A, Vásquez DH, Gatica H, Guerrero J. El tratamiento inmunomodulador precoz se relaciona con mejores resultados visuales en un subgrupo de pacientes con enfermedad de Vogt-Koyanagi-Harada. Acta Ophthalmol 2015; 93 (6): e475-480. https://PubMed.gov/25565265. DOI: 10.1111 / aos.12648
- Read RW, Rechodouni A, Butani N, Johnston R, LaBree LD, Smith RE, Rao NA. Complicaciones y factores pronósticos en la enfermedad de Vogt-Koyanagi-Harada. Am J Ophthalmol 2001; 131 (5): 599-606. https://PubMed.gov/11336934
Formato de cita sugerido
Mai AP, Tran C, Wilson CW, Fox AR, Boldt HC. Enfermedad de Vogt-Koyanagi-Harada (VKH). EyeRounds.org. 1 de abril de 2019. Disponible en http://EyeRounds.org/cases/284-vogt-koyanagi-harada.htm
Leave a Reply