Vogt-Koyanagi-Harada (VKH) -Krankheit
Autoren: Anthony P. Mai, BS; Charlene Tran, BS; Caroline W. Wilson, MD; Austin R. Fox, MD; H. Culver Boldt, MD
1. April 2019
ERSTPRÄSENTATION
Hauptbeschwerde
Verschwommenes Sehen und Kopfschmerzen
Geschichte der gegenwärtigen Krankheit/h3>
Eine 44-jährige Vietnamesin wurde der Notaufnahme mit einer 10-tägigen Vorgeschichte von progressivem verschwommenem Sehen in beiden Augen und einer dreitägigen Vorgeschichte von starken Kopfschmerzen vorgestellt. Ihr zentraler Sehverlust hatte sich mit einer Refraktion durch ihren Augenarzt nicht verbessert. Ihre schweren okzipitalen Kopfschmerzen verschlechterten sich mit der Bewegung und waren mit allgemeinem Unwohlsein, extremer Müdigkeit, leichter Photophobie und Tränenfluss verbunden. Paracetamol linderte die Schmerzen teilweise.
Sie war kürzlich nach Vietnam gereist, bestritt jedoch, dort kranke Kontakte geknüpft zu haben. Sie bestritt Kieferklaudikationen, Fieber oder Gewichtsveränderungen. Sie leugnete Hautausschläge, Hörveränderungen, Tinnitus, Schwindel, Taubheitsgefühl oder Kribbeln. Sie bestritt, jemals an Tuberkulose erkrankt zu sein. Sie hatte keine Vorgeschichte von Sehproblemen, Autoimmunerkrankungen oder Krebs.
Vorgeschichte des Auges
- Vorgeschichte einer kosmetischen Augenlidoperation (bilaterale Blepharoplastik) drei Jahre zuvor
- Keine Vorgeschichte eines Traumas oder einer Erkrankung des Auges
Vorgeschichte
Keine
Medikamente
Paracetamol nach Bedarf
Allergien
Keine bekannten Arzneimittelallergien
Familiengeschichte
Keine Vorgeschichte von Augenkrankheiten oder Autoimmunerkrankungen
Sozialgeschichte
Sie wanderte einige Jahre vor der Präsentation aus Vietnam aus. Sie ist verheiratet und hat drei Kinder. Sie arbeitet in einem Nagelstudio. Sie konsumiert keine Tabakprodukte, Alkohol oder illegale Substanzen. Sie reist alle sechs bis zwölf Monate nach Vietnam.
Überprüfung der Systeme
Negativ mit Ausnahme dessen, was in der Geschichte der gegenwärtigen Krankheit detailliert ist
AUGENUNTERSUCHUNG
Sehschärfe mit/ohne Korrektur (Snellen)
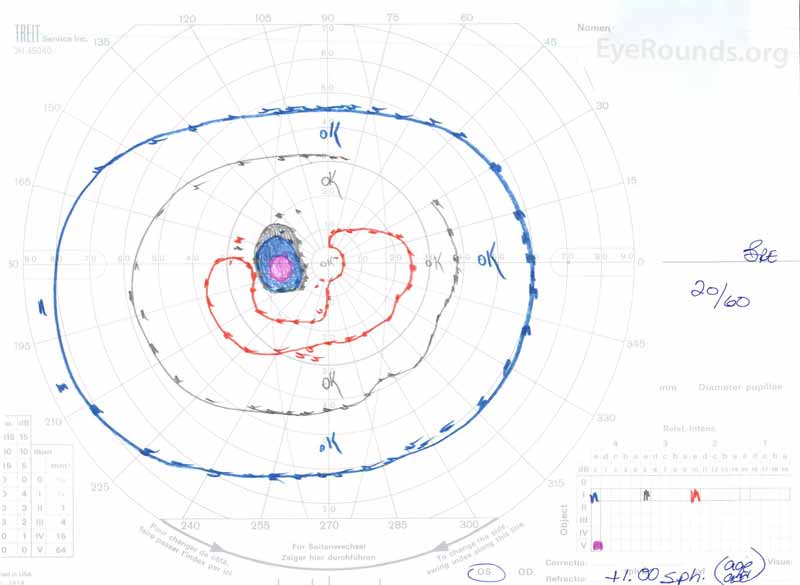
- Rechtes Auge (OD): 20/300 (keine Verbesserung mit Lochblende)
- Linkes Auge (OS): 20/60-2+2 (keine Verbesserung mit Pinhole)
Augenmotilität/Ausrichtung
Volle extraokulare Bewegungen in beiden Augen (OU)
Augeninnendruck (IOD): (Tonopen)
- OD: 12 mmHg
- OS: 14 mmHg
Pupillen
- OD: 4 mm im Dunkeln, 3 mm im Licht, kein relativ afferenter Pupillendefekt (RAPD)
- OS: 4 mm im Dunkeln, 3 mm im Licht, kein RAPD
)
- OD: Zentrales Skotom
- OS: Totaler inferotemporaler Defekt
Extern
Beidseitig normal
Spaltlampenuntersuchung
- Augenlider/Wimpern: Normal OU
- Bindehaut/Sklera: Klar und leise OU
- Hornhaut: 1+ punktförmige epitheliale Erosionen, keine keratischen Ausfällungen OU
- Vorderkammer: Spurenzelle und Flare und tief OU
- Iris: Normale Architektur OU
- Linse: Klar OU
Erweiterte Fundusuntersuchung (DFE)
- Glaskörper: Spur vordere Glaskörperzellen OU
- Scheibe:
- OD: Grad 3 bandscheibenödem, hyperämisch
- OS: Bandscheibenödem Grad 2-3, hyperämisch
- Cup-to-Disc-Verhältnis: 0,0 OU
- Makula:
- OD: 3+ zystoides Makulaödem (CME) und subretinale Flüssigkeit (SRF), die sich von der Bandscheibe bis zur temporalen Makula erstrecken. Kein Lipid oder Exsudate. Sumpfig erscheinende Aderhaut.
- OS: 2+ CME und SRF, die sich von der Scheibe durch die Fovea erstrecken. 1-2+ lineares Lipid, das sich von der Scheibe in Richtung Fovea erstreckt. Sumpfig erscheinende Aderhaut.
- Gefäße:
- OD: Zeitlich umhüllen
- OS: Normal
- Peripherie:
- OD: Zystischer Netzhautbüschel vor dem Äquator um 10:30 Uhr
- OS: Flacher SRF vor dem Äquator um 4:00
Abbildung 1: Farbige Fundusfotografien bei der Präsentation: (Linkes Bild) Das rechte Auge weist ein Bandscheibenödem und eine leichte Hyperämie sowie subretinale Flüssigkeit auf, die sich von der Bandscheibe temporal durch die Makula erstreckt. Es gibt auch eine fokale seröse Netzhautablösung superotemporal zur Bandscheibe entlang der oberen Arkade. (Right image) The left eye has disc edema and mild hyperemia, along with subretinal fluid extending from the disc to the macula and linear lipid deposits in the nasal macula.
 |
 |
 |
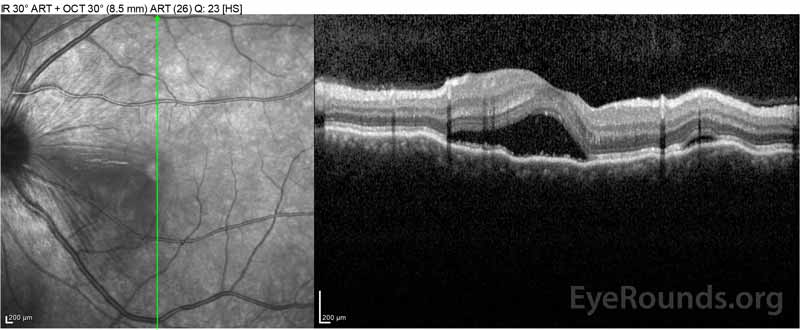 |
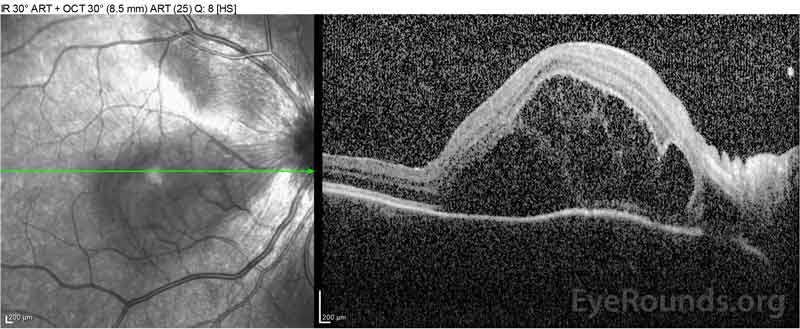
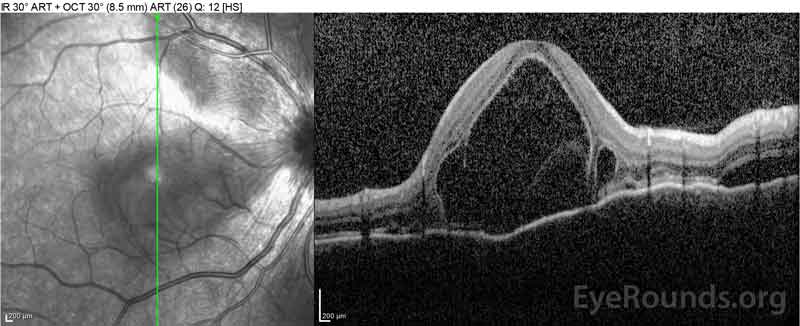
Figure 2: Die optische Kohärenztomographie (OCT) des rechten Auges (obere Platten) zeigt eine seröse Netzhautablösung der Fovea mit ausgedehnter darüberliegender intraretinaler Flüssigkeit, Störung der äußeren Netzhautschichten und Wellen der verdickten Aderhaut. OCT des linken Auges (unten) zeigt eine seröse Netzhautablösung in der nasalen Makula, die sich bis zur Fovea erstreckt.
 |
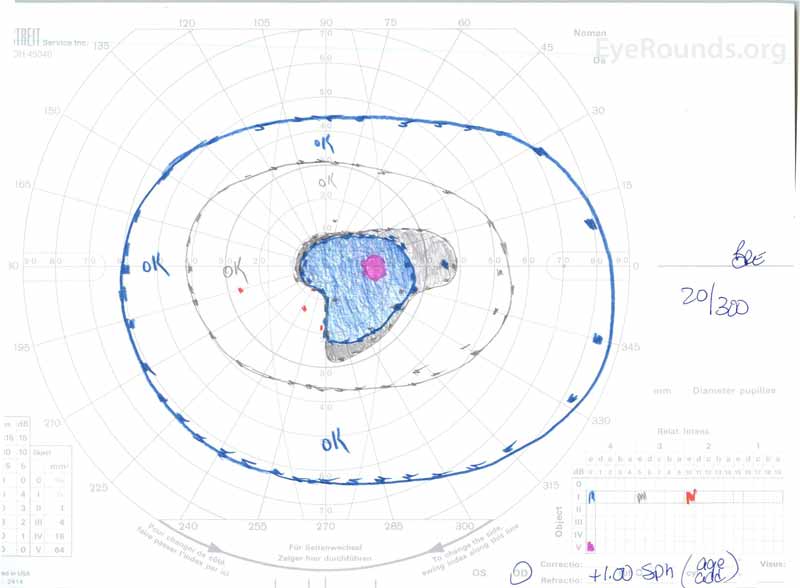 |
Abbildung 3: Goldman visuelle Felder (GVF), OU. (Linkes Bild) OS zeigt einen vergrößerten physiologischen blinden Fleck und ein mildes zentrales Skotom. (Rechtes Bild) OD zeigt ein mäßig dichtes zentrales Skotom, das den physiologischen blinden Fleck enthält und sich inferotemporal erstreckt.
B-Scan: Keine Anzeichen von Skleritis, leichte Glaskörpertrübungen/Zellen minderwertig
Differentialdiagnose
- Akute posteriore multifokale placoide Pigmentepitheliopathie (APMPPE)
- Zentrale seröse Chorioretinopathie
- Optikusneuritis
- Panuveitis
- Autoimmunerkrankung (z. B. SLE, Sarkoidose)
- Infektion (e.Syphilis, Tuberkulose, Bartonella henselae)
- Malignität (z. B. okuläres Lymphom)
- Posteriore Skleritis
- Sympathische Ophthalmie
- Uvealerguss-Syndrom
- Vogt-Koyanagi-Harada-Syndrom
AUFARBEITUNG
Vollständiges Blutbild
Anzahl der weißen Blutkörperchen: 4,9 K/ mm3 (Ref: 3,7-10,5)
Anzahl der roten Blutkörperchen 3,99 M/mm3 (Ref: 4,0-5,2)
Hämoglobin 11,6 g/ dl (Ref: 11,9-15,5)
Hämatokrit 35 % (Ref:: 35-47)
Basic metabolic panel
Sodium 138 mEq/L (Ref: 135-145)
Potassium 4.3 mEq/L (Ref: 3.5-5.0)
Chloride 107 mEq/L (Ref: 95-107)
CO2 20 mEq/L (Ref: 22-29)
Blood urea nitrogen 16 mEq/dL (Ref: 10-20)
Creatinine 0.7 mg/dL (Ref: 0.5-1.0)
C-reactive Protein (CRP): <0.5 mg/dL (Ref: <=0.5)
Erythrocyte sedimentation rate (ESR): 12 mm/Hr (Ref: 0-20)
Angiotensin–converting enzyme (ACE): 13 U/L (Ref: 8-52)
QuantiFERON-TB Gold: negativ
Eisen, Blut 54 Mikrogramm/ dl (Ref: 37-145)
Gesamteisenbindungskapazität 379 Mikrogramm/dl (Ref: 250-425)
KLINISCHER VERLAUF
Die Patientin wurde aufgrund ihrer Beschwerden über neu auftretende starke Kopfschmerzen und Sehverlust zunächst von der Notaufnahme untersucht. Gehirn-Computertomographie (CT) und Magnetresonanztomographie (MRT) -Scans waren unauffällig. ESR und CRP lagen innerhalb normaler Werte. Die Augenklinik untersuchte sie am nächsten Tag und stellte bilaterale seröse Netzhautablösungen und Panuveitis fest. ACE und QuantiFERON-TB Gold Labs waren beide negativ. Sie wurde mit Vogt-Koyanagi-Harada-Krankheit diagnostiziert, basierend auf ihrer klinischen Präsentation und asiatischer Abstammung. Sie wurde täglich mit 80 mg Prednison, Paracetamol bei Kopfschmerzen sowie Vitamin D und Kalzium behandelt. Ihre Kopfschmerzen ließen schnell nach und ihre Sehschärfe verbesserte sich in den folgenden zwei Wochen stetig. Ihre Prednison-Dosis wurde dann über drei Wochen auf 40 mg reduziert, wobei die Symptome weiterhin abklingen und die Sehschärfe verbessert wurde. Sie hatte kein Wiederauftreten von Kopfschmerzen oder Verschlechterung des Sehvermögens während der Prednison-Verjüngung. Bei ihrem letzten Termin hatte sie jeden zweiten Tag auf 5 mg abgenommen, ohne dass die Symptome wieder auftraten. Ihre Sehschärfe bei diesem Follow-up-Besuch betrug 20/15-2 OD und 20/20 + 2 OS, und die Makula-OCT zeigte eine vollständige Auflösung von Bandscheibenödemen und serösen Netzhautablösungen in beiden Augen (Abbildung 4).
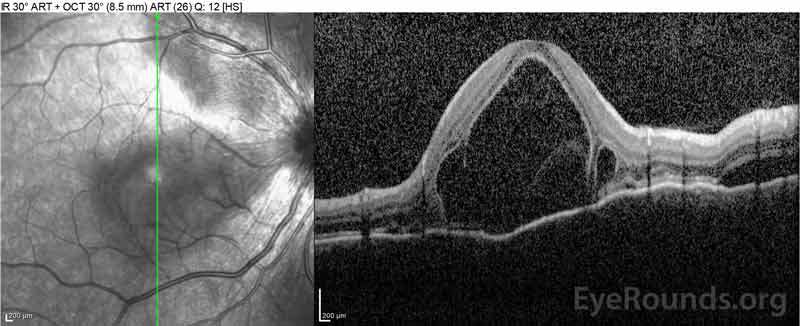
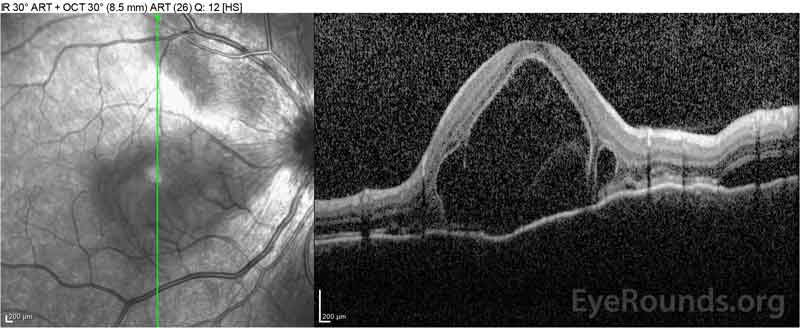
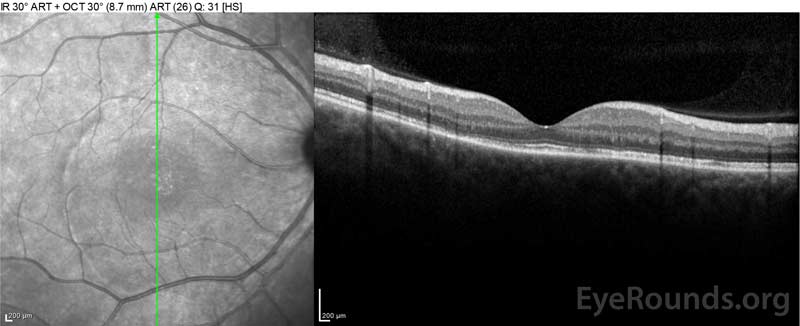
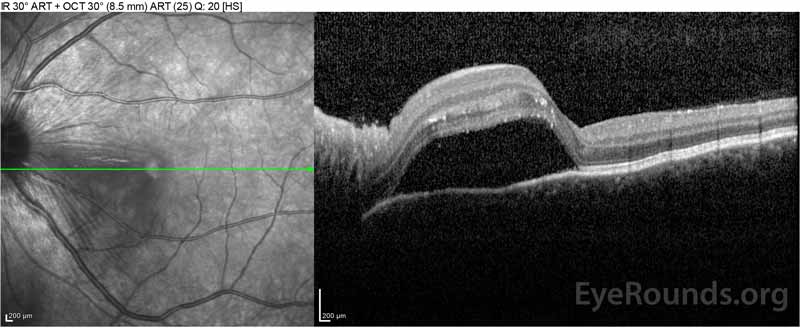
Abbildung 4: Optische Kohärenztomographie zeigt subretinale Flüssigkeit zu Studienbeginn (oben) und den Verlauf der Auflösung nach einer Woche (Mitte) und fünf Wochen (unten) während einer hochdosierten oralen Prednison-Verjüngung. Beachten Sie die Glättung der choroidalen Wellen mit der Behandlung.
 |
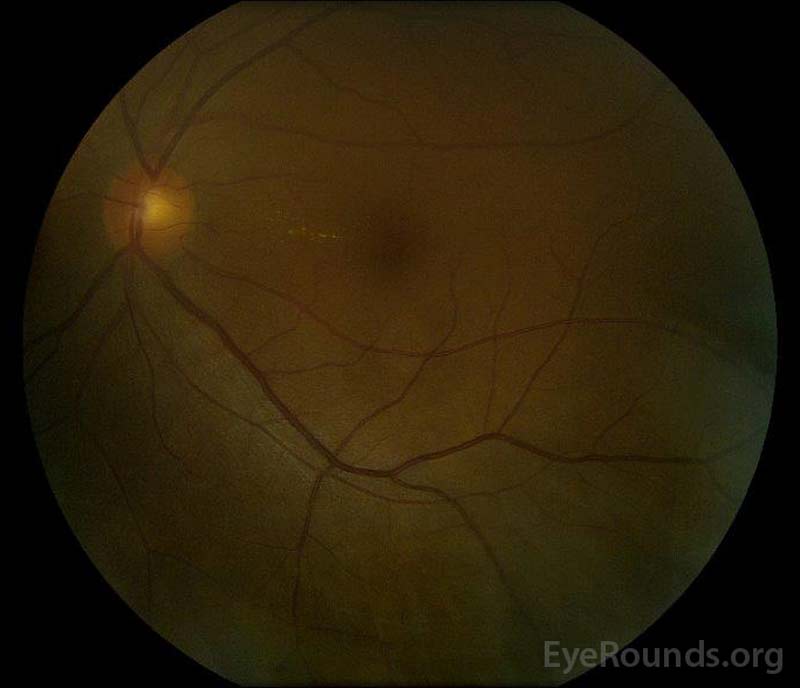 |
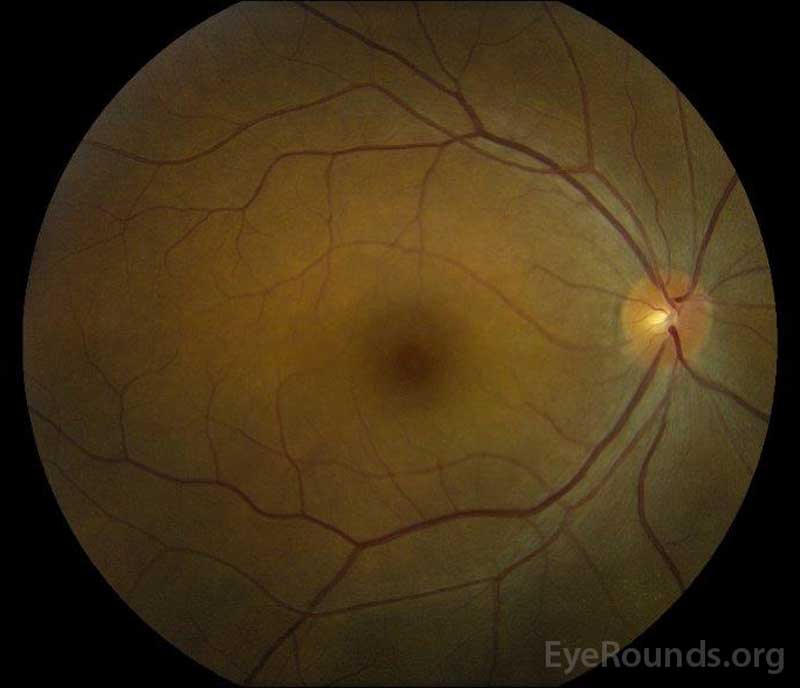
Abbildung 5: Farbfundusfotografie des rechten (A) und linken (B) Auges während der Rekonvaleszenzphase, die eine Verbesserung der subretinalen Flüssigkeit und des Bandscheibenödems zeigt.
DIAGNOSE
Unvollständige Vogt-Koyanagi-Harada-Krankheit
DISKUSSION
Die Vogt-Koyanagi-Harada-Krankheit (VKH) ist eine systemische Autoimmunerkrankung, die durch bilaterale nicht nekrotisierende granulomatöse Panuveitis gekennzeichnet ist, die mit extraokularen Integumentveränderungen wie Poliose und Vitiligo und Entzündungen der Uvea, des Innenohrs, der Haare und der Hirnhäute einhergeht. Harada-Krankheit ist die isolierte Uveitis ohne die systemischen Anzeichen oder Symptome von VKH.
Ätiologie
Die Ätiologie der VKH-Krankheit ist trotz aktueller Forschungsanstrengungen noch weitgehend unbekannt. Es wird angenommen, dass es sich um eine erworbene Autoimmunerkrankung handelt, die eine T-Zell-vermittelte Überempfindlichkeit gegen melanozytäre Selbstantigene mit einer zugrunde liegenden genetischen Prädisposition und einem möglichen mikrobiellen Auslöser beinhaltet . Tyrosinase und Tyrosinase-verwandte Peptide sind Melanozytenantigene, die als Ziele von Autoimmunprozessen in VKH vorgeschlagen wurden . Ein erhöhtes Risiko für eine VKH-Erkrankung war jedoch laut einer Studie nicht mit der Tyrosinase-Genfamilie assoziiert .
Aufgrund der erhöhten Prävalenz bei bestimmten ethnischen und geschlechtsspezifischen Gruppen wird angenommen, dass eine genetische Prädisposition in der Pathogenese von VKH vorliegt. Mehrere Gene, einschließlich humaner Leukozytenantigen (HLA) und Interleukin (IL) Gene, wurden mit VKH in verschiedenen ethnischen Populationen assoziiert . HLA-Rezeptoren sind wichtige Histokompatibilitätskomplexe beim Menschen, die dem Immunsystem Peptide präsentieren. HLA-DR1, HLA-DR4, HLA-DRB1 * 0405 und HLA-DRw53 sind mehrere Haplotypen, die bei Patienten mit VKH-Krankheit gefunden werden . HLA-DR4 tritt häufiger bei Japanern und Hispanics auf, während HLA-DRB1 * 0405 häufiger bei koreanischen und nahöstlichen Patienten auftritt . Sowohl die HLA-DR4- als auch die HLA-DRB1 * 0405-Allele werden bei vietnamesischen Patienten gefunden . Der HLA-DRB1-Rezeptor bindet in unterschiedlichen Kapazitäten an Melanozytenantigene. Trotz dieser Assoziationen werden Gentests derzeit nicht empfohlen.
Angesichts der üblichen Prodromalsymptome, die mit VKH einhergehen, einschließlich Fieber, Kopfschmerzen, Meningismus und Tinnitus, wurde eine anregende virale Ätiologie als Auslöser für das Auftreten von VKH durch Mechanismen der molekularen Mimikry bei genetisch prädisponierten Patienten vorgeschlagen. Cytomegalovirus-Hüllglykoprotein H weist eine signifikante Aminosäurehomologie zum Tyrosinase-Peptid auf, und eine CMV-Infektion kann VKH durch molekulare Mimikry auslösen (d. H. Erkennung durch HLA-Klasse-II-Rezeptoren) . Ebstein-Bar-Virus (EBV) wurde ebenfalls in Verbindung gebracht. Es gab jedoch keine definitiven Hinweise auf eine virale Ätiologie von VKH, und es bleibt unklar, was die VKH-Autoimmunantwort auslöst .
Pathophysiologie
Es gibt vier klassische Phasen der VKH, die unterschiedliche Darstellungen haben können: prodromal, akut uveitisch, rekonvaleszent und chronisch rezidivierend. Histopathologische Veränderungen beginnen typischerweise in der akuten Phase .
Die akute uveitische Phase ist durch eine bilaterale Verdickung der Uvea infolge einer granulomatösen Entzündung gekennzeichnet. Die Granulome bestehen aus Lymphozyten, Makrophagen und granulatgefüllten Epithel- und Riesenzellen . Obwohl bisher angenommen wurde, dass es sich bei den Epitheloidzellen um veränderte Melanozyten handelt, schlug eine immunhistochemische Follow-up-Studie stattdessen einen Ursprung aus Gewebemakrophagen vor . Granulome, die mit epitheloiden Histiozyten gefüllt sind und als Dalen-Fuchs-Knötchen bezeichnet werden, sind häufig zwischen dem retinalen Pigmentepithel (RPE) und der Bruch-Membran zu sehen. Die granulomatöse Entzündung der Uvea führt zu einer Verdickung der Aderhaut und exsudativen Netzhautablösungen, die mit Proteinflüssigkeit gefüllt sind. Darüber hinaus ist die Pleozytose (i.e., erhöhte Zellzahl) kann in der Vorderkammer und Glaskörper vorhanden sein.
Die Rekonvaleszenzphase wird durch Depigmentierung der Aderhaut und der extraokularen Bereiche einschließlich der periokularen Haut und der Haare identifiziert. Eine depigmentierte Aderhaut vor einem blassen Sehnerv vermittelt den Eindruck eines „Sunset-Glow“ -Fundus, der ein klassisches Merkmal dieser Phase von VKH ist . Darüber hinaus treten Dalen-Fuchs-Knötchen in der Rekonvaleszenzphase unter dem RPE stärker auf .
Die chronisch-rezidivierende Phase ist gekennzeichnet durch verminderte Aderhautdicke, Auflösung seröser Netzhautablösungen, chronische leichte Vitritis und rezidivierende granulomatöse Entzündung des vorderen Segments. Choroidale Neovaskularisation (CNV) und subretinale Fibrose können sich während dieser Phase entwickeln und sind Indikatoren für ein schweres Fortschreiten der Krankheit . Katarakte und sekundäres Glaukom sind weitere Komplikationen einer langjährigen oder wiederkehrenden Entzündung in dieser Phase .
Epidemiologie
VKH ist bei Rassen mit dunklerem Hautpigment vorherrschend, insbesondere bei Asiaten, Südamerikanern, Menschen aus dem Nahen Osten und amerikanischen Ureinwohnern. Die VKH-Krankheit macht >10% der Uveitis in diesen Populationen aus . Es wird angenommen, dass nur 1-4% der Uveitis-Fälle sekundär zur VKH-Krankheit in den Vereinigten Staaten sind (7). In den Vereinigten Staaten wurde festgestellt, dass die meisten Fälle von VKH Personen asiatischer, hispanischer und / oder indianischer Abstammung betreffen . Interessanterweise betrifft die VKH-Krankheit Afrikaner trotz ihrer dunklen Pigmentierung selten . Die Inzidenz der VKH-Krankheit variiert stark zwischen den Rassenuntergruppen in den Nachbarländern . Zum Beispiel ist Koreas Inzidenz von VKH nur 2%, viel niedriger als in Japan und China . VKH hat einen typischen Beginn im Alter von 20 bis 50 Jahren ; Studien deuten jedoch darauf hin, dass 3,1-13,4% der VKH-Fälle pädiatrische Patienten sind und 10% der Fälle ≥65 Jahre alt sind . Klassisch, Es wird angenommen, dass VKH eine Vorliebe für das weibliche Geschlecht hat, und während die meisten Studien zeigen, dass VKH Frauen überproportional betrifft, Einige Studien haben eine männliche Veranlagung oder keine geschlechtsspezifische Veranlagung gezeigt .
Anzeichen / Symptome
Wie bereits erwähnt, sind die vier Stadien der VKH-Krankheit prodromal, uveitisch, rekonvaleszent und chronisch rezidivierend. Jedes Stadium weist unterschiedliche klinische Merkmale auf. Prodromal: Dieses Anfangsstadium kann sich als grippeähnliche Erkrankung mit überwiegend konstitutionellen Symptomen wie Kopfschmerzen, Schwindel, Fieber, Müdigkeit und/ oder Übelkeit darstellen. Neurologische Symptome von Meningitis, Hirnnervenlähmung und Optikusneuritis sowie auditive Symptome von Tinnitus, Dysakusis und Schwindel wurden berichtet . Photophobie, verschwommenes Sehen, Floater und / oder Augenschmerzen beginnen normalerweise innerhalb von 48 Stunden nach Prodromalsymptomen . Die Prodromalphase dauert typischerweise einige Tage bis Wochen.
Diagnosekriterien
Die neuesten Diagnosekriterien, die sogenannten Revised Diagnostic Criteria (RDC) für VKH, wurden 1999 auf dem Ersten Internationalen Workshop zu VKH definiert . Diese sind in Tabelle 1 dargestellt. Die RDC sind insofern nützlich, als sie VKH in drei verschiedene diagnostische Kategorien einteilen, basierend auf der Krankheitsphase, in der sich ein Patient präsentiert: vollständig, unvollständig und wahrscheinlich. Diese Kategorisierung der Krankheit ermöglicht eine angemessene und frühzeitige Behandlung der „wahrscheinlichen“ Krankheit, die dazu beitragen kann, das Fortschreiten der „vollständigen“ Krankheit zu verhindern.
Die Aufarbeitung anderer Ursachen für Augenentzündungen, sowohl infektiöser als auch autoinflammatorischer Art, ist unerlässlich. Dazu gehören Erythrozytensedimentationsrate (ESR), C-reaktives Protein (CRP), Quantiferon-Gold-Tests auf Tuberkulose, Rapid Plasma Reagin (RPR) für Syphilis, Angiotensin-Converting-Enzym (ACE) und eine Röntgenaufnahme des Brustkorbs für Sarkoidose, antinukleäre Antikörper (ANA) und p- / c-ANCA. Außerdem muss eine Vorgeschichte eines kürzlichen Okulartraumas oder einer intraokularen Operation notiert werden und deutet wahrscheinlich auf eine sympathische Ophthalmie (SO) als wahrscheinlichere Diagnose hin, da die Darstellung und Pathophysiologie zwischen SO und VKH sehr ähnlich ist .
Um eine Diagnose von VKH in zweideutigen Fällen zu unterstützen, kann eine Lumbalpunktion durchgeführt werden, um nach lymphozytischer und monozytischer Pleozytose zu suchen; Dies wird jedoch klinisch selten angewendet. Achtzig Prozent der Patienten haben innerhalb einer Woche eine Pleozytose in der Liquor cerebrospinalis (CSF) und 97% innerhalb von drei Wochen eine Pleozytose. Erhöhte Spiegel von Immunzellen können bis zu acht Wochen nach Ausbruch der Krankheit anhalten . Die T-Zell-Oberflächenmarkerprofile sind zwischen dem Liquor und dem Kammerwasser ähnlich, unterscheiden sich jedoch vom Blut. Dies deutet auf die Fähigkeit von CSF hin, die Entzündung der Uvea bei der VKH-Krankheit genau zu reflektieren .
Tabelle 1. Überarbeitete diagnostische Kriterien für die Vogt-Koyanagi-Harada-Krankheit
*Aus Tabelle 1 in (15).
„Vollständige Vogt-Koyanagi-Harada-Krankheit (Kriterien 1 bis 5 müssen vorliegen)
- Keine Vorgeschichte eines penetrierenden Okulartraumas oder einer Operation vor dem ersten Auftreten einer Uveitis.
- Keine klinischen oder Laborbeweise, die auf andere Augenerkrankungen hindeuten.
- Bilaterale Augenbeteiligung (a oder b müssen erfüllt sein, abhängig vom Krankheitsstadium bei der Untersuchung des Patienten).
- Frühe Manifestationen der Krankheit.
- Es muss eine diffuse Choroiditis (mit oder ohne anteriore Uveitis, Glaskörperentzündungsreaktion oder Papillenhyperämie)vorliegen, die sich als eine der folgenden Manifestationen manifestieren kann:
- Frühe Manifestationen der Krankheit.
- Fokale Bereiche der subretinalen Flüssigkeit oder
- Bullöse seröse Netzhautablösungen.
- Mit zweideutigen Fundusbefunden; Beide der folgenden Befunde müssen ebenfalls vorliegen:
- Fokale Bereiche mit Verzögerung der Aderhautperfusion, multifokale Bereiche mit punktueller Leckage, große placoide Bereiche mit Hyperfluoreszenz, Bündelung in subretinaler Flüssigkeit und Färbung des Sehnervs (in der Reihenfolge des sequentiellen Auftretens aufgeführt) durch Fluoreszeinangiographie und
- Diffuse Verdickung der Aderhaut, ohne Anzeichen einer posterioren Skleritis durch Ultraschall.
- Späte Manifestationen der Krankheit.
- Anamnese, die auf frühere Befunde aus 3a hindeutet, und entweder beide (2) und (3) unten oder mehrere Anzeichen aus (3):
- Augendepigmentierung (eine der folgenden Manifestationen ist ausreichend): (a) Sunset Glow Fundus oder (b) Sugiura-Zeichen.
- Andere Augenzeichen:
- Nummuläre chorioretinale depigmentierte Narben oder
- Verklumpung und/oder Migration des Pigmentepithels der Netzhaut oder
- Rezidivierende oder chronische Uveitis anterior.
- Neurologische /auditive Befunde (können zum Zeitpunkt der Untersuchung abgeklungen sein).
- Meningismus (Unwohlsein, Fieber, Kopfschmerzen, Übelkeit, Bauchschmerzen, Steifheit des Nackens und des Rückens oder eine Kombination dieser Faktoren; Kopfschmerzen allein reichen jedoch nicht aus, um die Definition von Meningismus zu erfüllen) oder
- Tinnitus oder
- Pleozytose der Liquor cerebrospinalis.
- Integumentärer Befund (nicht vor Beginn des Zentralnervensystems oder der Augenerkrankung).
- Alopezie oder
- Poliose oder
- Vitiligo.
Unvollständige Vogt-Koyanagi-Harada-Krankheit (Kriterien 1 bis 3 und entweder 4 oder 5 müssen vorliegen)
- Keine Vorgeschichte eines penetrierenden Okulartraumas oder einer Operation vor dem anfänglichen Auftreten einer Uveitis und
- Keine klinischen oder Laborbeweise, die auf andere okuläre Erkrankungen hindeuten Entitäten und
- Bilaterale Augenbeteiligung.
- Neurologische/auditive Befunde; wie oben für die vollständige Vogt-Koyanagi-Harada-Krankheit definiert, oder
- Integumentäre Befunde; wie oben für die vollständige Vogt-Koyanagi-Harada-Krankheit definiert.
Wahrscheinliche Vogt-Koyanagi-Harada-Krankheit (isolierte Augenerkrankung; Kriterien 1 bis 3 müssen vorliegen)
- Keine Vorgeschichte eines penetrierenden Okulartraumas oder einer Operation vor dem ersten Auftreten einer Uveitis.
- Keine klinischen oder Laborbeweise, die auf andere Augenerkrankungen hindeuten.
- Bilaterale Augenbeteiligung, wie oben für die vollständige Vogt-Koyanagi-Harada-Krankheit definiert. „
Testing/Laboratory Work-up
Bei der ersten Aufarbeitung von VKH sollten folgende Tests in Betracht gezogen werden:
- Optische Kohärenztomographie (OCT): In der akuten uveitischen Phase zeigt OCT wahrscheinlich eine signifikante Verdickung der Aderhaut und seröse Netzhautablösungen. Die subretinalen Flüssigkeitsansammlungen können Septationen aufweisen, von denen angenommen wird, dass sie Fibrinmembranen und Entzündungsprodukte sind, wodurch eine lobuläre Struktur entsteht, die auch bei der Fluoreszeinangiographie zu sehen ist. In der Rekonvaleszenzphase kann OCT Bereiche der Netzhautverdünnung nach gelöster Entzündung nach Kortikosteroidbehandlung nachweisen .
- B-Scan Ultraschall: In der akuten Phase kann die Sonographie eine diffuse Verdickung der hinteren Aderhaut, eine Verdickung der hinteren Sklera, Netzhautablösungen und Glaskörpertrübungen zeigen . Ziliarergüsse können mit Ultraschall-Biomikroskopie beobachtet werden . Dieser Test ist auch nützlich, um eine posteriore Skleritis auszuschließen.
- Fluoreszeinangiographie (FA): Klassisch zeigt die FA in der Frühphase multifokale choroidale hypofluoreszente Punkte, gefolgt von multiplen fokalen hyperfluoreszenten Bereichen mit diffuser Leckage in der Spätphase . Der Farbstoff tritt durch das RPE aus und sammelt sich im subretinalen Raum, der die hyperfluoreszierenden Punkte umgibt. FA kann diagnostisch nützlich sein, wenn die VKH-Krankheit ohne extraokulare Symptome auftritt. Papillenhyperfluoreszenz und Fensterdefekte, die durch atrophische chorioretinale Narben verursacht werden, können in der mittleren Peripherie beobachtet werden . FA im chronisch-rezidivierenden Stadium der VKH-Erkrankung zeigt unspezifische Fensterdefekte aufgrund von RPE-Schäden, choroidaler Neovaskularisation und subretinaler Fibrose .
- Indocyaningrün (ICG) Angiographie: ICG in der Frühphase zeigt hyperfluoreszierende Stromagefäße, die auf eine Aderhautvaskulopathie hinweisen, und hypofluoreszierende dunkle Punkte, die Granulomen und einer verzögerten fleckigen Füllung des Aderhautgefäßsystems entsprechen . Die späte Phase zeigt unscharfe stromale Gefäßmuster und diffuse Aderhauthyperfluoreszenz. Die Hyperfluoreszenz der Bandscheibe deutet auf eine schwere Erkrankung hin. ICGA kann subklinische Aderhautentzündungen in sehr frühen Stadien oder sogar nach systemischer Therapie erkennen .
- Lumbalpunktion: Pleozytose in der Liquor cerebrospinalis ist bei der Mehrzahl der VKH-Patienten vorhanden. Die Lumbalpunktion sollte früh im Krankheitsverlauf durchgeführt werden, da sich die Pleozytose auflösen kann
Behandlung / Management / Richtlinien
Die Behandlungsziele bei VKH umfassen die Früherkennung und Unterdrückung aktiver Entzündungen sowie die Prävention wiederkehrender Entzündungen und sehbedrohlicher Komplikationen wie Glaukom, bullöser Netzhautablösung und Aderhautneovaskularisation.
Die systemische Kortikosteroidbehandlung ist die bevorzugte Therapie für die VKH-Krankheit, insbesondere während des akuten uveitischen Stadiums. Es wurde gezeigt, dass der Verabreichungsweg von Kortikosteroiden (oral versus intravenös) die Sehschärfe oder das Auftreten visuell signifikanter Komplikationen bei der Behandlung von akutem VKH nicht beeinflusst . Bei schweren Erkrankungen ist das vorgeschlagene Protokoll die intravenöse Verabreichung von Methylprednisolon für drei Tage, gefolgt von einer oralen hochdosierten Prednisonbehandlung. Bei leichten bis mittelschweren Erkrankungen kann hochdosiertes orales Prednison mit 1-2 mg / kg / Tag ausreichend sein. Die Steroiddosis sollte über einen Zeitraum von etwa sechs Monaten langsam verringert werden, um ein erneutes Auftreten zu verhindern . Eine aggressive frühzeitige Behandlung kann zusammen mit seriellen FA-Tests, die das Verschwinden von Farbstofflecks durch das RPE zeigen, dazu beitragen, ein weiteres Fortschreiten der Krankheit, ein Wiederauftreten und extraokulare Manifestationen zu verhindern . Topische Steroide und Zykloplegika können Zellen in der Vorderkammer und im Glaskörper verringern.
Intravitreale und Sub-Tenon-Injektionen von Triamcinolon wurden zur kurzfristigen Kontrolle der intraokularen Entzündung während der akuten oder rezidivierenden Phasen verwendet; diese lokalen Therapien sollten bei widerspenstigen Erkrankungen und bei Patienten in Betracht gezogen werden, die die ungünstigen systemischen Nebenwirkungen von Steroiden aufgrund der verlängerten Steroidverjüngung schlecht vertragen. Intravitreale Anti-VEGF-Injektionen werden manchmal zur Kontrolle der choroidalen Neovaskularisation und bei anhaltenden fovealen serösen Netzhautablösungen eingesetzt . Steroidsparende Mittel, einschließlich Antimetaboliten, Calcineurin-Inhibitoren, Biologika, TNF-alpha-Inhibitoren oder zytotoxische Mittel, können zur Behandlung von VKH verwendet werden und sollten sorgfältig überwacht werden, häufig in Abstimmung mit einem rheumatologischen Dienst . Es gab anhaltende Diskussionen über die Verwendung von nichtsteroidalen Immunsuppressiva als Erstlinientherapie für die VKH-Krankheit. Eine kürzlich durchgeführte Studie ergab jedoch keine Unterschiede in den Ergebnissen zwischen der frühen immunmodulatorischen Erstlinientherapie (IMT) und der Prednison-Behandlung allein . Darüber hinaus sind immunsuppressive und biologische Therapien teuer und erfordern eine sorgfältige Bewertung der Vorbehandlung sowie eine häufige Nachuntersuchung mit Blutuntersuchungen, um schwerwiegende Nebenwirkungen festzustellen.
Im chronisch-rezidivierenden Stadium kann ein häufiges Wiederauftreten auf eine Resistenz gegen eine Kortikosteroidtherapie hindeuten und auf die Notwendigkeit einer steroidsparenden immunmodulatorischen Behandlung hindeuten . Das bevorzugte Mittel bei steroidresistentem Rezidiv oder Steroidunverträglichkeit ist Cyclosporin . Infliximab, Rituximab, Adalimumab und Interferon alpha-2a sind biologische Wirkstoffe, die auch zur Behandlung von refraktärer Uveitis bei VKH-Erkrankungen eingesetzt wurden.
Zur Behandlung der anterioren Uveitis, die häufig mit akutem VKH einhergeht, sollten je nach Grad der Vorderkammerentzündung topische Steroide (z. B. Prednisolonacetat 1%) und topische Zyloplegie (z. B. Cyclopentolat 1% oder Atropin 1%) verschrieben werden.
Augenkomplikationen sind häufig mit der VKH-Krankheit verbunden. Angesichts der verschiedenen Stadien und der Vielfalt der Präsentationen, in denen ein Patient mit VKH auftreten kann, kann sich die Behandlung in vielen Fällen verzögern. Bei schweren Formen von VKH und bei Rezidiven kann eine intraokulare Entzündung schwer zu kontrollieren sein und zu strukturellen Schäden führen. Über 50% der Patienten entwickeln damit verbundene Komplikationen, einschließlich Katarakt, sekundärem Glaukom, choroidalen neovaskulären Membranen, subretinaler Fibrose oder einer Kombination davon (Abbildung 6) .
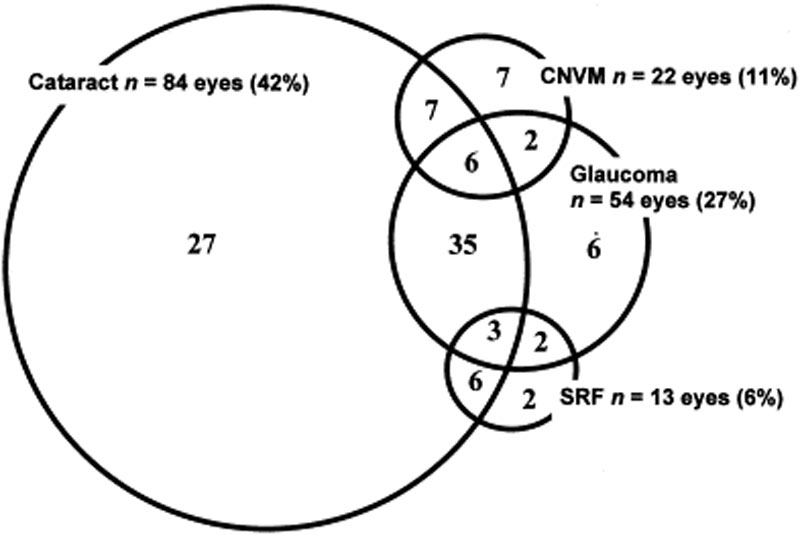
Abbildung 6: Venn-Diagramm mit Komplikationen bei VKH-Patienten. (Verwendet mit Genehmigung von Am J Ophthalmol. 2001;131(5):599-606 )
EPIDEMIOLOGIE UND ÄTIOLOGIE
|
SIGNS
|
SYMPTOME
|
BEHANDLUNG/MANAGEMENT
|
- Du L, Kijlstra A, Yang P. Vogt-Koyanagi-Harada-Krankheit: Neue Einblicke in Pathophysiologie, Diagnose und Behandlung. Prog Retin Auge Res 2016;52:84-111. https://PubMed.gov/26875727. Geburtsdatum: 10.1016/j.preteyeres.2016.02.002
- Yamaki K, Gocho K, Hayakawa K, Kondo I, Sakuragi S. Proteine der Tyrosinase-Familie sind Antigene, die für die Vogt-Koyanagi-Harada-Krankheit spezifisch sind. J Immunol 2000;165(12):7323-7329. https://PubMed.gov/11120868
- Horie Y., Takemoto Y., Miyazaki A., Namba K., Kase S., Yoshida K., Ota M., Hasumi Y., Inoko H., Mizuki N., Ohno S. Tyrosinase-Genfamilie und Vogt-Koyanagi-Harada-Krankheit bei japanischen Patienten. Mol Vis 2006;12:1601-1605. https://PubMed.gov/17200659
- Ng JY, Luk FO, Lai TY, Pang KP. Einfluss der Molekulargenetik auf die Vogt-Koyanagi-Harada-Krankheit. J Ophthalmic Inflamm Infizieren 2014;4:20. https://PubMed.gov/25097674. Artikelnummer: 10.1186/s12348-014-0020-1
- Bowling B. Uveitis. Kanskis klinische Augenheilkunde New York, New York: Elsevier; 2016; Kapitel 11; p. 395-465.
- Yeh PT YC, Yang CH, Lin CP. Nichtrhegmatogene Netzhautablösung. In: Schachat AP SS, Hinton DR, Wilkinson CP, Wiedemann P,, Herausgeber. Ryans Netzhaut. New York: Elsevier; 2018; Kapitel 99; p. 1828-1849.
- Goto H RK, Rao N. Vogt–Koyanagi–Harada-Krankheit. In: Schachat AP SS, Hinton DR, Wilkinson CP, Wiedemann P, Herausgeber. Ryans Netzhaut. New York, New York: Elsevier; 2018; Kapitel 78; p. 1505-1515.
- Riddington L, Hall AJ, Tait B, Nicholson I, Varney M. Vogt-Koyanagi-Harada-Syndrom bei Patienten vietnamesischer Abstammung. Aust NZ J Ophthalmol 1996;24(2):147-149. https://PubMed.gov/9199747
- Sugita S, Takase H, Kawaguchi T, Taguchi C, Mochizuki M. Kreuzreaktion zwischen Tyrosinase-Peptiden und Cytomegalovirus-Antigen durch T-Zellen von Patienten mit Vogt-Koyanagi-Harada-Krankheit. Int Ophthalmol 2007;27(2-3):87-95. https://PubMed.gov/17253112. UST-IDNR.: 10.1007/s10792-006-9020- y
- Freund BK SD, Mieler WF, Yannuzzi LA. Entzündung. Der Netzhautatlas. New York, New York: Elsevier 2017; Kapitel 4; p. 279-398.
- Rao N. Vogt-Koyanagi-Harada-Krankheit. In: J YMaD, Herausgeber. Augenheilkunde. New York, New York: Elsevier; 2014; Kapitel 7.17; p. 761-763.
- Rao NA, Xu S, Font RL. Sympathische Ophthalmie. Eine immunhistochemische Untersuchung von Epitheloid- und Riesenzellen. Augenheilkunde 1985;92 (12): 1660-1662. https://PubMed.gov/4088616
- Nussenblatt RB. Vogt-Koyanagi-Harada-Syndrom. In: Zeitschrift für Soziologie und Soziologie. Uveitis: Grundlagen und klinische Praxis. 4. Auflage ed: Elsevier; 2010; Kapitel Kapitel 24.Lesen Sie RW, Holland GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L, Pivetti-Pezzi P, Tessler HH, Usui M. Überarbeitete diagnostische Kriterien für die Vogt-Koyanagi-Harada-Krankheit: Bericht eines internationalen Komitees für Nomenklatur. Am J Ophthalmol 2001;131(5):647-652. https://PubMed.gov/11336942
- Chung H, Choi DG. Klinische Analyse von Uveitis. Korean J Ophthalmol 1989;3(1):33-37. https://PubMed.gov/2795939. Ursprungsbezeichnung: 10.3341/kjo.1989.3.1.33
- Abu El-Asrar AM, Al-Kharashi AS, Aldibhi H, Al-Fraykh H, Kangave D. Vogt-Koyanagi-Harada-Krankheit bei Kindern. Auge (Lond) 2008;22(9):1124-1131. https://PubMed.gov/17479116. Ursprungsbezeichnung: 10.1038/sj.Auge.6702859
- Martin TD, Rathinam SR, Cunningham ET al. Prävalenz, klinische Merkmale und Ursachen des Sehverlusts bei Kindern mit Vogt-Koyanagi-Harada-Krankheit in Südindien. Retina 2010;30(7):1113-1121. https://PubMed.gov/20168275. Ursprungsbezeichnung: 10.1097/IAE.0b013e3181c96a87. Einseitige Manifestation des Vogt-Koyanagi-Harada-Syndroms bei einem 7-jährigen Kind. Am J Ophthalmol 1991;111(3):380-382. https://PubMed.gov/2000916
- Yamamoto Y, Fukushima A, Nishino K, Koura Y, Komatsu T, Ueno H. Vogt-koyanagi-Harada-Krankheit mit Beginn bei älteren Patienten im Alter von 68 bis 89 Jahren. Jpn J Ophthalmol 2007;51(1):60-63. https://PubMed.gov/17295144. UST-IDNR.: 10.1007/s10384-006-0379-0
- Wang Y, Chan CC. Geschlechtsspezifische Unterschiede bei der Vogt-Koyanagi-Harada-Krankheit und der sympathischen Ophthalmie. J Ophthalmol 2014;2014:157803. https://PubMed.gov/24734166. DOI: 10.1155/2014/157803
- Nakao K, Abematsu N, Mizushima Y, Sakamoto T. Papillenschwellung bei Vogt-Koyanagi-Harada-Krankheit. Invest Ophthalmol Vis Sci 2012;53(4):1917-1922. https://PubMed.gov/22408010. Kennziffer: 10.1167/iovs.11-8984
- Rao NA, Gupta EIN, Dustin L, Chee SP, Okada AA, Khairallah M, Bodaghi B, Lehoang P, Accorinti M, Mochizuki M, Prabriputaloong T, Lesen RW. Häufigkeit der Unterscheidung klinischer Merkmale bei der Vogt-Koyanagi-Harada-Krankheit. Ophthalmology 2010;117(3):591-599, 599.e591. https://PubMed.gov/20036008. DOI: 10.1016/j.ophtha.2009.08.030
- Veerappan M, Fleischman D, Ulrich JN, Stinnett SS, Jaffe GJ, Allingham RR. The Relationship of Vogt-Koyanagi-Harada Syndrome to Ocular Hypertension and Glaucoma. Ocul Immunol Inflamm 2017;25(6):748-752. https://PubMed.gov/27438521. DOI: 10.1080/09273948.2016.1189578
- Baltmr A, Lightman S, Tomkins-Netzer O. Vogt-Koyanagi-Harada syndrome – current perspectives. Clin Ophthalmol 2016;10:2345-2361. https://PubMed.gov/27932857. DOI: 10.2147/OPTH.S94866
- Kitaichi N, Matoba H, Ohno S. Die positive Rolle der Lumbalpunktion bei der Diagnose der Vogt-Koyanagi-Harada-Krankheit: Lymphozytenuntergruppen im Kammerwasser und in der Liquor cerebrospinalis. Int Ophthalmol 2007;27(2-3):97-103. https://PubMed.gov/17211585. UST-IDNR.: 10.1007/s10792-006-9016-7
- Oshima Y, Harino S, Hara Y, Tano Y. Angiographische Befunde von Indocyaningrün bei Vogt-Koyanagi-Harada-Krankheit. Am J Ophthalmol 1996;122(1):58-66. https://PubMed.gov/8659599
- Lesen Sie RW, Yu F, Accorinti M, Bodaghi B, Chee SP, Fardeau C, Goto H, Holland GN, Kawashima H, Kojima E, Lehoang P, Lemaitre C, Okada AA, Pivetti-Pezzi P, Secchi A, Siehe RF, Tabbara KF, Usui M, Rao NA. Bewertung der Auswirkungen auf die Ergebnisse des Verabreichungsweges von Kortikosteroiden bei akuter Vogt-Koyanagi-Harada-Krankheit. Am J Ophthalmol 2006;142(1):119-124. https://PubMed.gov/16815259. Geburtsdatum: 10.1016/j.ajo.049.02.2006
- Rubsamen PE, Gass JD. Vogt-Koyanagi-Harada-Syndrom. Klinischer Verlauf, Therapie und langfristiges visuelles Ergebnis. Arch Ophthalmol 1991;109(5):682-687. https://PubMed.gov/2025171
- Urzua CA, Velasquez V, Sabat P, Berger O, Ramirez S, Goecke A, Vásquez DH, Gatica H, Guerrero J. Eine frühere immunmodulatorische Behandlung ist bei einer Untergruppe von Patienten mit Vogt-Koyanagi-Harada-Krankheit mit besseren visuellen Ergebnissen verbunden. Acta Ophthalmol 2015;93(6):e475-480. https://PubMed.gov/25565265. Zertifikate: 10.1111/aos.12648
- Lesen Sie RW, Rechodouni A, Butani N, Johnston R, LaBree LD, Smith RE, Rao NA. Komplikationen und prognostische Faktoren bei der Vogt-Koyanagi-Harada-Krankheit. Am J Ophthalmol 2001;131(5):599-606. https://PubMed.gov/11336934
Vorgeschlagenes Zitierformat
Mai AP, Tran C, Wilson CW, Fox AR, Boldt HC. Vogt-Koyanagi-Harada (VKH) -Krankheit. EyeRounds.org . 1. April 2019. Verfügbar ab http://EyeRounds.org/cases/284-vogt-koyanagi-harada.htm
Leave a Reply