Vogt-Koyanagi-Harada (VKH) sjukdom
författare: Anthony P. Mai, BS; Charlene Tran, BS; Caroline W. Wilson, MD; Austin R. Fox, MD; H. Culver Boldt, MD
1 April 2019
inledande PRESENTATION
huvudklagomål
suddig syn och huvudvärk
historia av nuvarande sjukdom
en 44-årig Vietnamesisk kvinna presenterade för akutavdelningen med en 10-dagars historia av progressiv suddig syn i båda ögonen och en tre dagars historia av svår huvudvärk. Hennes centrala synförlust hade inte förbättrats med en brytning av hennes optiker. Hennes svåra occipitala huvudvärk förvärrades med rörelse och var förknippade med generaliserad sjukdom, extrem trötthet, mild fotofobi och riva. Paracetamol lindrade delvis smärtan.
hon hade nyligen rest till Vietnam men nekade att möta sjuka kontakter där. Hon förnekade käftklaudikering, feber eller viktförändringar. Hon förnekade hudutslag, hörselförändringar, tinnitus, yrsel, domningar eller stickningar. Hon förnekade någonsin att ha tuberkulos. Hon hade ingen historia av tidigare synproblem, autoimmuna tillstånd eller cancer.
tidigare okulär historia
- historia av kosmetisk ögonlockskirurgi (bilateral blepharoplasty) tre år tidigare
- ingen historia av okulärt trauma eller sjukdom
tidigare sjukdomshistoria
ingen
läkemedel
paracetamol vid behov
allergier
Inga kända läkemedelsallergier
familjehistoria
ingen historia av ögonsjukdom eller autoimmun sjukdom
social historia
hon invandrade från Vietnam flera år före presentationen. Hon är gift och har tre barn. Hon arbetar på en nagelsalong. Hon konsumerar inte tobaksprodukter, alkohol eller olagliga ämnen. Hon reser till Vietnam var sjätte till tolv månad.
granskning av system
negativ förutom vad som beskrivs i historien om nuvarande sjukdom
okulär undersökning
synskärpa med / utan korrigering (Snellen)
- höger öga (OD): 20/300 (ingen förbättring med pinhole)
- vänster öga (OS): 20/60-2+2 (ingen förbättring med pinhole)
okulär motilitet/justering
fulla extraokulära rörelser i båda ögonen (OU)
intraokulärt tryck (IOP): (Tonopen)
- OD: 12 mmHg
- OS: 14 mmHg
elever
- OD: 4 mm i mörker, 3 mm i ljus, ingen relativ afferent pupilldefekt (RAPD)
- OS: 4 mm i mörker, 3 mm i ljus, ingen RAPD
konfrontation visuella fält: (räkna H3>
- od: Central scotoma
- OS: Total inferotemporal defekt
extern
normal på båda sidor
spaltlampa examen
- lock/fransar: normal ou
- konjunktiva/sclera: klar och tyst ou
- hornhinna: 1+ punktera epiteliala erosioner, inga keratiska fällningar OU
- främre kammare: Spårcell och flare och djup OU
- Iris: Normal arkitektur OU
- lins: klar OU
utvidgad fundusundersökning (DFE)
- glaskropp: spåra främre glaskroppsceller OU
- skiva:
- OD: grad 3 skivödem, hyperemisk
- OS: grad 2-3 skivödem, hyperemisk
- kopp-till-skivförhållande: 0,0 ou
- makula:
- od: 3+ cystoid makulärt ödem (CME) och subretinalvätska (SRF) som sträcker sig från skivan till den temporala makulaen. Inga lipid eller exsudater. Boggy-förekommer choroid.
- OS: 2 + CME och SRF sträcker sig från skivan genom fovea. 1-2 + linjär lipid som sträcker sig från skiva mot fovea. Boggy-förekommer choroid.
- fartyg:
- OD: mantel temporärt
- OS: Normal
- periferi:
- OD: cystisk retinal tuft anterior till ekvatorn vid 10: 30
- OS: Grunt SRF anterior till ekvatorn vid 4:00
 |
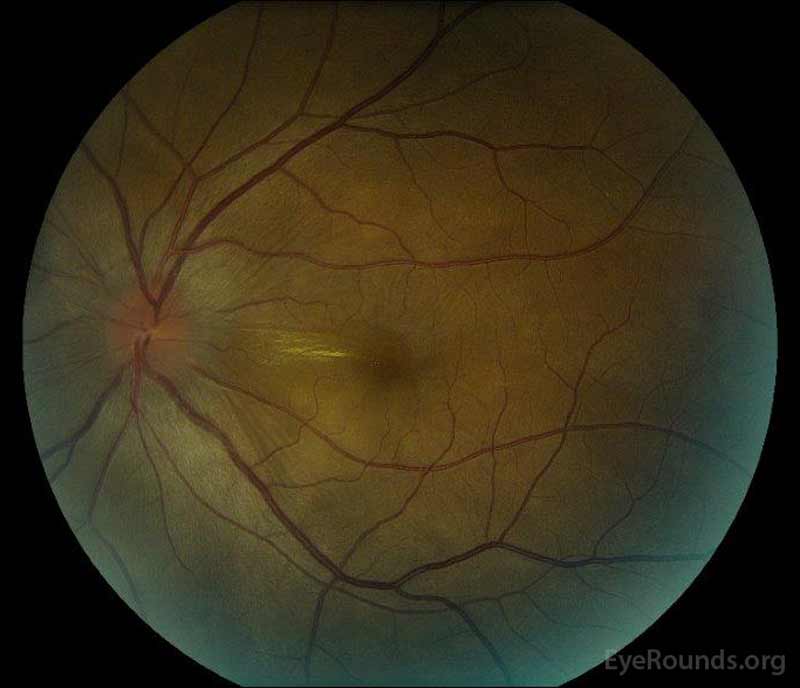 |
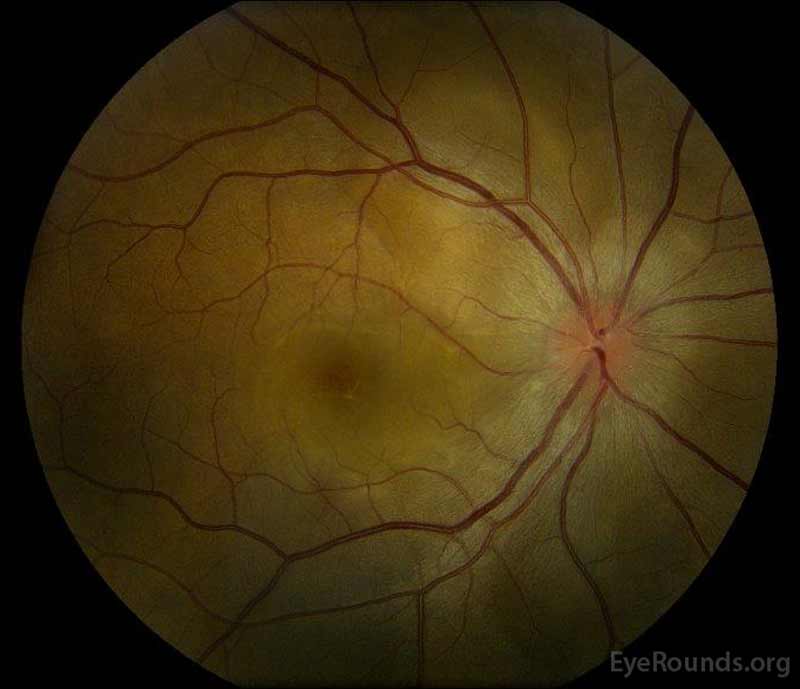
figur 1: färg fundusfotografier vid presentation: (vänster bild) det högra ögat har skivödem och mild hyperemi samt subretinalvätska som sträcker sig från skivan temporärt genom makula. Det finns också en fokal serös näthinneavlossning superotemporal till skivan, längs den överlägsna arkaden. (Right image) The left eye has disc edema and mild hyperemia, along with subretinal fluid extending from the disc to the macula and linear lipid deposits in the nasal macula.
 |
 |
 |
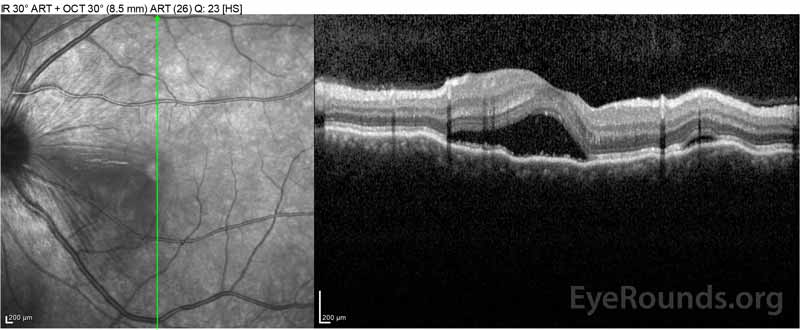 |
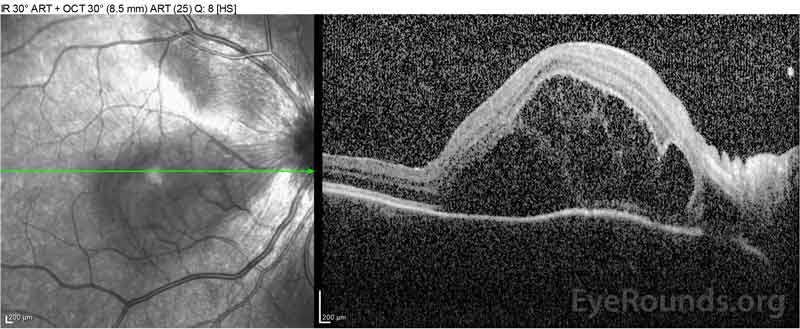
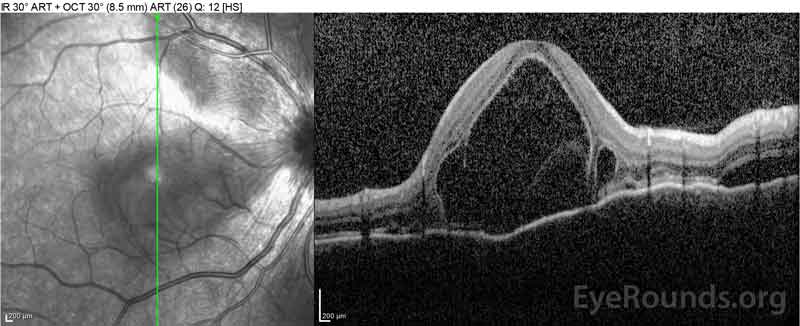
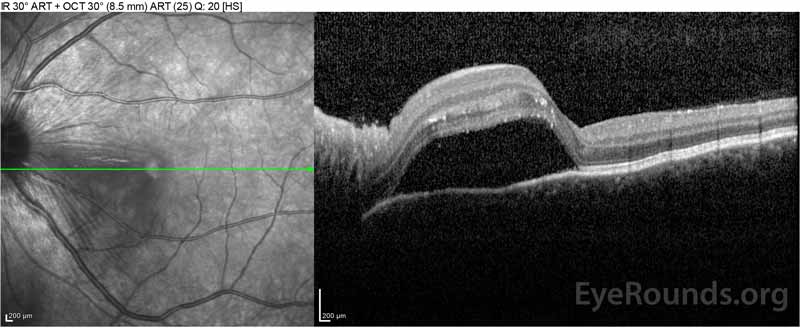
Figure 2: Optisk koherens tomografi (OCT) i höger öga (övre paneler) visar en serös näthinneavlossning som involverar fovea med omfattande överliggande intraretinalvätska, störning av de yttre retinala skikten och vågningar av den förtjockade koroiden. OCT av vänster öga (bottenpaneler) visar en serös näthinneavlossning i näsmakula som sträcker sig upp till fovea.
 |
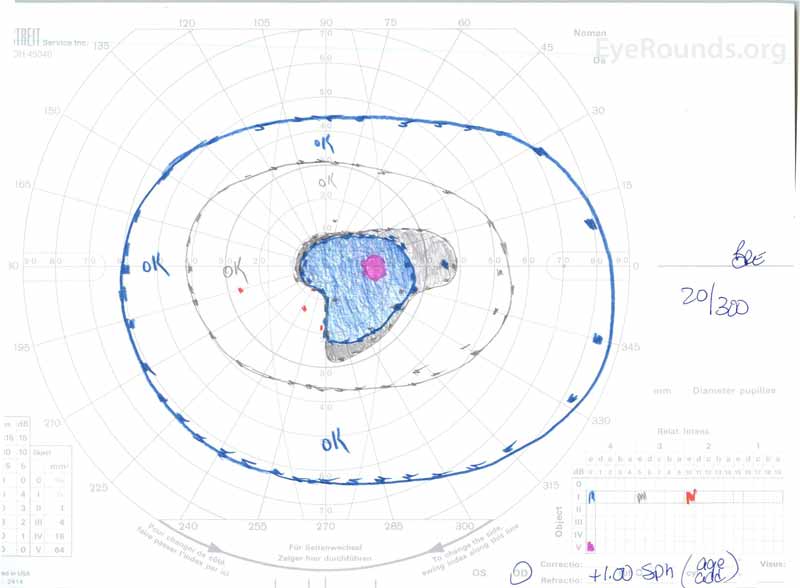 |
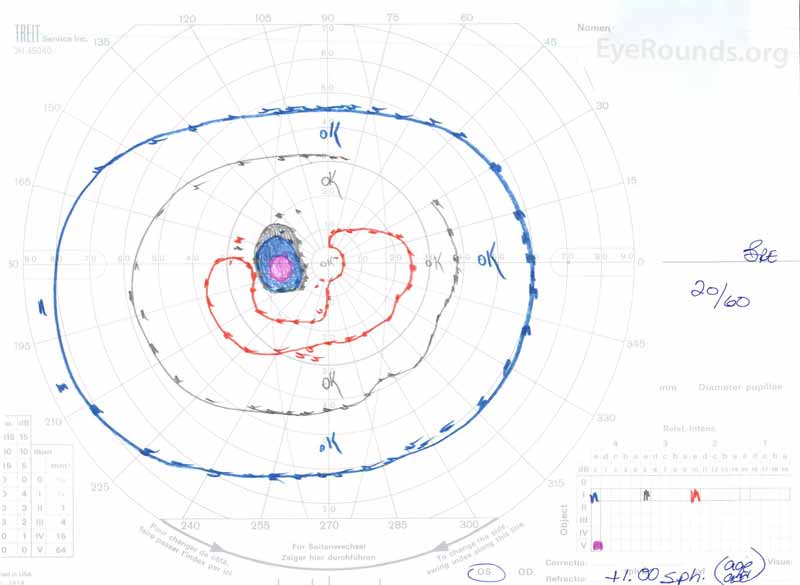
figur 3: Goldman visuella fält (GVF), OU. (Vänster bild) OS visar en förstorad fysiologisk blind fläck och mild Central scotoma. (Höger bild) od visar ett måttligt tätt centralt scotom som innehåller den fysiologiska blinda fläcken och sträcker sig inferotemporalt.
B-scan: inga tecken på sklerit, milda vitreala opaciteter/celler inferiorly
differentialdiagnos
- akut bakre multifokal placoidpigmentepiteliopati (APMPPE)
- Central serös korioretinopati
- optisk neurit
- Panuveit
- autoimmun sjukdom (t.ex. SLE, sarkoidos)
- infektion (e.g., syfilis, tuberkulos, Bartonella henselae)
- malignitet (t. ex. okulärt lymfom)
- bakre sklerit
- sympatisk oftalmi
- Uveal effusionssyndrom
- Vogt-Koyanagi-Haradas syndrom
WORK-UP
fullständigt blodtal
antal vita blodkroppar: 4,9 k/mm3 (ref: 3,7-10,5)
antal röda blodkroppar 3,99 m/mm3 (ref: 4,0-5,2)
hemoglobin 11,6 g/dl (Ref: 11,9-15,5)
hematokrit 35 % (ref:: 35-47)
Basic metabolic panel
Sodium 138 mEq/L (Ref: 135-145)
Potassium 4.3 mEq/L (Ref: 3.5-5.0)
Chloride 107 mEq/L (Ref: 95-107)
CO2 20 mEq/L (Ref: 22-29)
Blood urea nitrogen 16 mEq/dL (Ref: 10-20)
Creatinine 0.7 mg/dL (Ref: 0.5-1.0)
C-reactive Protein (CRP): <0.5 mg/dL (Ref: <=0.5)
Erythrocyte sedimentation rate (ESR): 12 mm/Hr (Ref: 0-20)
Angiotensin–converting enzyme (ACE): 13 U/L (Ref: 8-52)
QuantiFERON-TB Gold: negativt
järn, blod 54 mikrogram / dL (Ref: 37-145)
Total järnbindningskapacitet 379 mikrogram/dL (Ref: 250-425)
klinisk kurs
patienten utvärderades initialt av akutmottagningen med tanke på hennes klagomål om allvarlig huvudvärk och synförlust. Hjärnberäknad tomografi (CT) och magnetisk resonansavbildning (MRI) skanningar var unremarkable. ESR och CRP var inom normala nivåer. Oftalmologikliniken utvärderade henne dagen efter och fann bilaterala serösa retinala avdelningar och panuveit. ACE och QuantiFERON-TB Gold labs var båda negativa. Hon diagnostiserades med Vogt-Koyanagi-Harada sjukdom baserat på hennes kliniska presentation och asiatisk härkomst. Hon behandlades med 80 mg prednison dagligen, acetaminophen efter behov för huvudvärk och vitamin D och kalciumtillskott. Hennes huvudvärk försvann snabbt och hennes synskärpa förbättrades stadigt under de följande två veckorna. Hennes prednisondosering avsmalnades sedan till 40 mg under tre veckor med fortsatt upplösning av symtom och förbättring av synskärpa. Hon hade ingen återkommande huvudvärk eller försämrad syn under prednison avsmalningen. Vid hennes senaste möte, hon hade avsmalnande ner till 5 mg varannan dag, utan återkomst av symtom. Hennes synskärpa vid det uppföljningsbesöket var 20/15-2 OD och 20/20+2 OS, och macular OCT visade full upplösning av skivödem och serösa retinala avdelningar i båda ögonen (Figur 4).
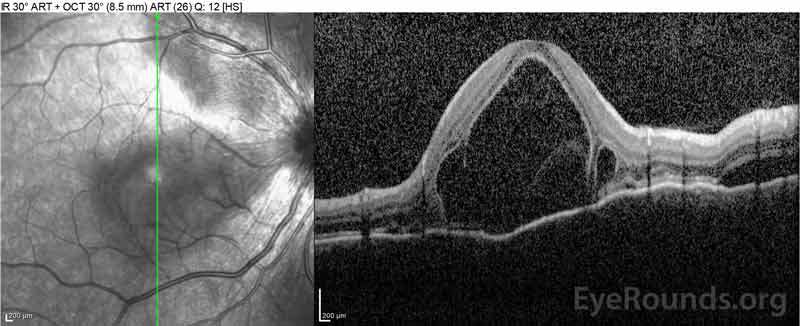
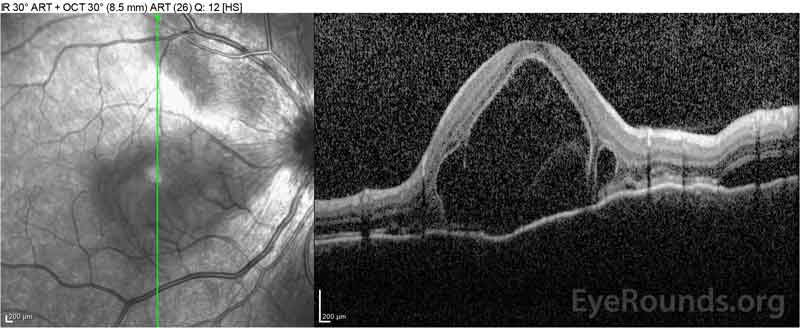
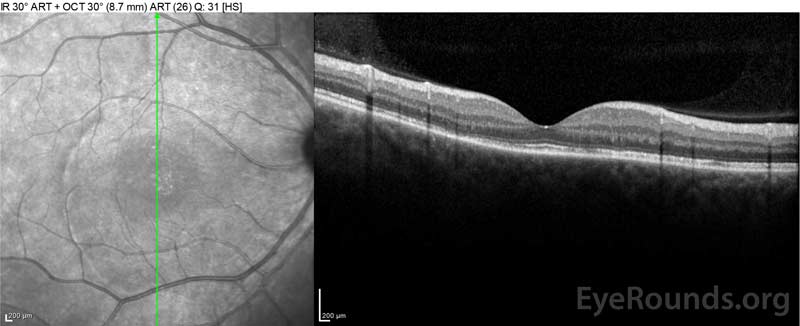
Figur 4: Optisk koherens tomografi som visar subretinalvätska vid baslinjen (topp) och upplösningen vid en vecka (mitten) och fem veckor (botten) medan den är på en högdos oral prednison avsmalning. Observera utjämningen av koroidala vågor med behandling.
 |
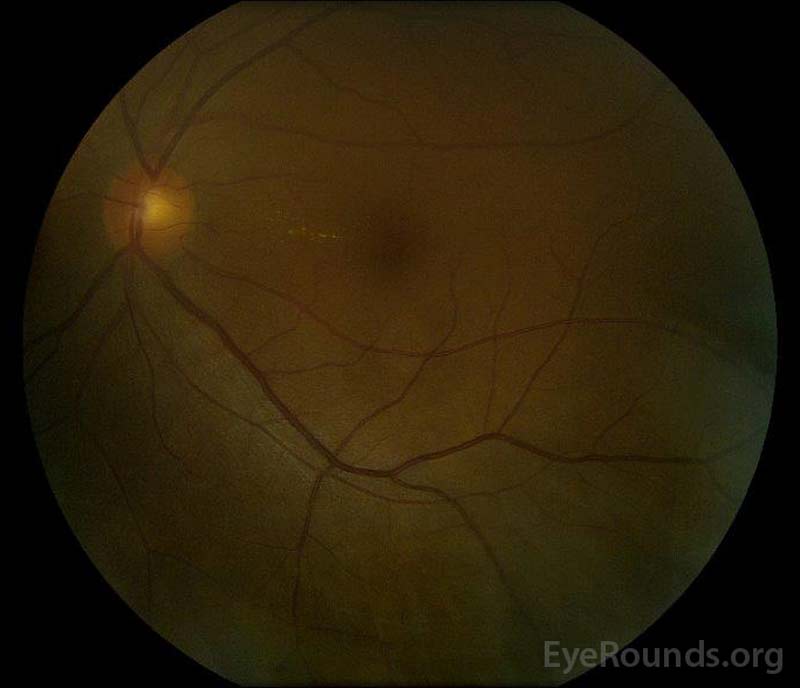 |
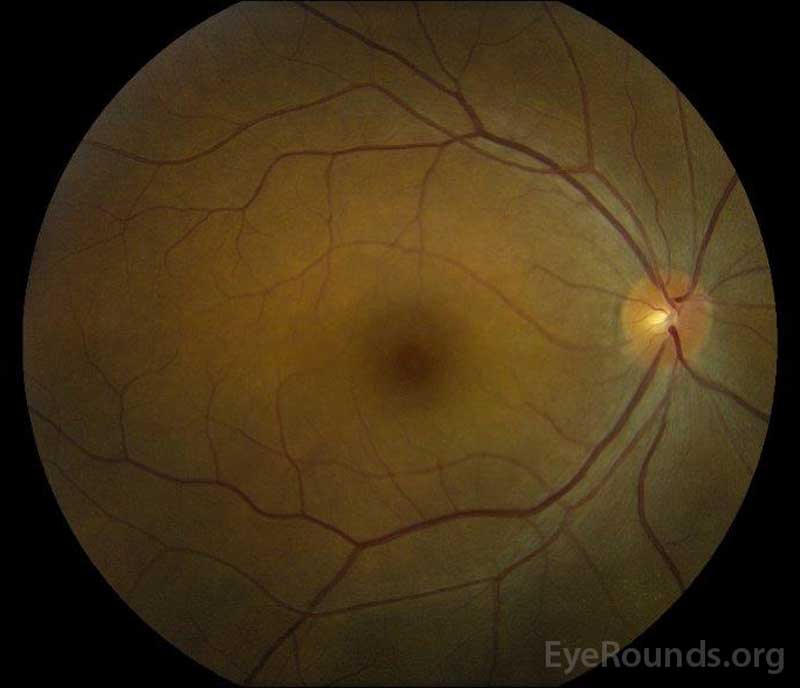
figur 5: Färg fundusfotografering av höger (a) och vänster (B) ögon Under konvalescensfasen som visar förbättring av subretinalvätska och skivödem.
diagnos
ofullständig Vogt-Koyanagi – Harada sjukdom
diskussion
Vogt-Koyanagi-Harada (VKH) sjukdom är ett systemiskt autoimmunt tillstånd som kännetecknas av bilateral icke-nekrotiserande granulomatös panuveit associerad med extraokulära integumentära förändringar, såsom polios och vitiligo, och inflammation som påverkar uvea, inre örat, hår och hjärnhinnor. Harada sjukdom är den isolerade uveit utan systemiska tecken eller symtom på VKH.
etiologi
etiologin för VKH-sjukdomen är fortfarande i stort sett okänd trots nuvarande forskningsinsatser. Det tros vara en förvärvad autoimmun sjukdom som involverar t-cellmedierad överkänslighet mot melanocytiska självantigener, med en underliggande genetisk predisposition och möjlig mikrobiell utlösare . Tyrosinas-och tyrosinasrelaterade peptider är melanocytantigener som har föreslagits som mål för autoimmuna processer i VKH . Ökad risk för VKH-sjukdom var emellertid inte associerad med tyrosinasgenfamiljen, enligt en studie .
På grund av den ökade prevalensen bland vissa etniska och könsgrupper tros det finnas en genetisk predisposition i patogenesen av VKH. Flera gener, inklusive humana leukocytantigen (HLA) och interleukin (IL) gener, har associerats med VKH i olika etniska populationer . HLA-receptorer är stora histokompatibilitetskomplex hos människor som presenterar peptider i immunsystemet. HLA-DR1, HLA-DR4, HLA-DRB1*0405 och HLA-DRw53 är flera haplotyper som finns hos patienter med VKH-sjukdom . HLA-DR4 är vanligare hos japanska och spansktalande människor, medan HLA-DRB1 * 0405 är vanligare hos koreanska och Mellanöstern patienter . Både HLA-DR4 och HLA-DRB1*0405 alleler finns hos vietnamesiska patienter . HLA-DRB1-receptorn binder till melanocytantigener i varierande kapacitet. Trots dessa föreningar rekommenderas inte genetisk testning vid denna tidpunkt.
Med tanke på de vanliga prodromala symtomen som åtföljer VKH, inklusive feber, huvudvärk, meningismus och tinnitus, har en anstiftande viral etiologi föreslagits som en utlösare för VKH-debut genom mekanismer för molekylär mimik hos genetiskt predisponerade patienter. Cytomegalovirus kuvert glykoprotein H har signifikant aminosyrahomologi till tyrosinaspeptiden, och CMV-infektion kan utlösa VKH genom molekylär mimikri (dvs erkännande av HLA-klass II-receptorer) . Ebstein-bar virus (EBV) har också varit inblandad. Det har emellertid inte funnits några definitiva bevis för en viral etiologi av VKH, och det är fortfarande oklart vad som utlöser VKH autoimmun respons .
patofysiologi
det finns fyra klassiska faser av VKH som kan ha variabla presentationer: prodromal, akut uveitisk, konvalescent och kronisk återkommande. Histopatologiska förändringar börjar vanligtvis i den akuta fasen .
den akuta uveitiska fasen kännetecknas av bilateral uvealförtjockning sekundär till granulomatös inflammation. Granulomerna består av lymfocyter, makrofager och granulfyllda epitelioid-och jätteceller . Även om epitelioidcellerna tidigare tros vara förändrade melanocyter, föreslog en uppföljning immunhistokemisk studie ett ursprung från vävnadsmakrofager istället . Granulom fyllda med epitelioida histiocyter, benämnda Dalen-Fuchs noduler, kan ofta ses mellan retinalpigmentepitelet (RPE) och Bruchs membran. Uveal granulomatös inflammation leder till koroidal förtjockning och exudativa retinala avdelningar fyllda med proteinhaltig vätska. Dessutom pleocytos (i.e., ökat cellantal) kan vara närvarande i den främre kammaren och glaskroppen .
den konvalescenta fasen identifieras genom depigmentering av koroid-och extraokulära områden, inklusive periokulär hud och hår. En depigmenterad koroid som sätts mot en blek optisk nerv ger intrycket av en” solnedgångsglöd ” fundus, vilket är ett klassiskt inslag i denna fas av VKH . Dessutom blir Dalen-Fuchs knölar mer framträdande under RPE i den konvalescent fasen .
den kronisk-återkommande fasen kännetecknas av minskad koroidal tjocklek, upplösning av serösa retinala avdelningar, kronisk mild vitrit och återkommande granulomatös främre segmentinflammation. Koroidal neovaskularisering (CNV) och subretinal fibros kan utvecklas under denna fas och är indikatorer på allvarlig sjukdomsprogression . Katarakt och sekundär glaukom är andra komplikationer av långvarig eller återkommande inflammation i denna fas .
epidemiologi
VKH är vanligt i raser med mörkare hudpigment, särskilt asiater, sydamerikaner, Mellanöstern och indianer. VKH-sjukdom står för > 10% av uveit i dessa populationer . Endast 1-4% av uveitfall anses vara sekundära till VKH-sjukdom i USA (7). I USA har de flesta fall av VKH visat sig påverka individer av Asiatisk, spansktalande och/eller Indian anständigt . Intressant nog påverkar VKH-sjukdomen sällan afrikaner trots deras mörka pigmentering . Förekomsten av VKH-sjukdom varierar kraftigt mellan rasundergrupper i grannländerna . Till exempel är Koreas förekomst av VKH bara 2%, mycket lägre än den som finns i Japan och Kina .
VKH har en typisk början på 20 till 50 års ålder ; studier tyder dock på att 3, 1-13, 4% av VKH-fallen är pediatriska patienter och 10% av fallen är 65-åringar . Klassiskt anses VKH ha en förkärlek för det kvinnliga könet, och medan de flesta studier visar att VKH oproportionerligt påverkar kvinnor, har några studier visat en manlig predisposition eller ingen könsfördelning .
tecken / symtom
som tidigare nämnts är de fyra stadierna av VKH-sjukdomen prodromala, uveitiska, konvalescent och kroniska återkommande. Varje steg uppvisar tydliga kliniska egenskaper.
- Prodromal: detta inledande skede kan uppvisa som en influensaliknande sjukdom med övervägande konstitutionella symtom, såsom huvudvärk, yrsel, feber, trötthet och / eller illamående. Neurologiska symtom på hjärnhinneinflammation, kranialnervpalsier och optisk neurit, liksom hörselsymtom på tinnitus, dysakus och svindel har rapporterats . Fotofobi, suddig syn, floaters och/eller ögonsmärta börjar vanligtvis inom 48 timmar efter prodromala symtom . Den prodromala fasen varar vanligtvis från några dagar till veckor.
- akut Uveitic: detta stadium inkluderar suddig syn, fotofobi, konjunktivalinjektion och ögonsmärta. Det kan finnas mild främre uveit som först verkar icke-granulomatös. Unilateral debut övergår vanligtvis till bilateralt engagemang inom 1-2 veckor. Granulomatös främre uveit med fårkött-fett keratiska fällningar kan utvecklas. Posteriora undersökningsfynd kan innefatta optisk nervödem och hyperemi , multifokala områden av koroidit, flera områden av serösa retinala avdelningar lokaliserade till den bakre fundus, koroidal förtjockning, utstrålande korioretinala veck och vitrit . Serösa näthinneavskiljningar kan bilda ett klöverbladig mönster i den bakre fundus och kan utvecklas till omfattande bullousavskiljningar i svåra fall . Akut inflammatorisk glaukom har associerats med denna fas av sjukdomen och kan uppvisa en grund främre kammare sekundär till ciliär kroppsödem, som efterliknar akut vinkelförslutning . Varaktigheten av den akuta uveitiska fasen beror på snabb diagnos och hantering.
- kronisk Uveitisk eller konvalescent: detta stadium utvecklas vanligtvis flera veckor efter den akuta fasen och kännetecknas av vitiligo (t .ex. ansikte, händer, axlar eller rygg), polios och alopeci. Depigmentering nära hornhinnans limbus, känd som Sugiuras tecken, kan ses en månad efter sjukdomsuppkomsten ; detta tecken ses dock sällan utanför den japanska befolkningen . Koroidal depigmentering sker vanligtvis under några månader och resulterar i den ljusa orangefärgade färgen på koroid och den klassiska ”sunset glow fundus.”Sunset glow fundus anses vara den viktigaste och prediktiva vid diagnosen kronisk VKH . Väldefinierade, runda, nummulära korioretinala ärr kan bildas i mitten av periferin. Den kroniska uveitiska fasen varar vanligtvis flera månader.
- kronisk-återkommande: Detta stadium kännetecknas av återkommande episoder av granulomatös främre uveit med fårkött fett keratiska fällningar, iris noduler, iris depigmentering, bakre synechiae, bakre subkapsulära grå starr, sekundär glaukom, koroidala neovaskulära membran, och i slutändan subretinal fibros och nummulär korioretinal atrofi . Den kroniska fasen utvecklas vanligtvis minst sex månader efter den första presentationen. De serösa näthinneavdelningarna som finns under de akuta och konvalescentfaserna återkommer vanligtvis inte med aggressiv kortikosteroidbehandling .
diagnostiska kriterier
de senaste diagnostiska kriterierna, med namnet reviderade diagnostiska kriterier (RDC) för VKH, definierades 1999 vid den första internationella workshopen om VKH . Dessa beskrivs i Tabell 1. RDC är användbara genom att de delar VKH i tre olika diagnostiska kategorier baserat på sjukdomsfasen under vilken en patient presenterar: fullständig, ofullständig och sannolik. Denna kategorisering av sjukdom möjliggör lämplig och tidig hantering av ”sannolik” sjukdom som kan bidra till att förhindra progression till ”fullständig” sjukdom.
arbete för andra orsaker till okulär inflammation, både infektiös och autoinflammatorisk, är väsentliga. Dessa kan inkludera erytrocytsedimenteringshastighet( ESR), C-reaktivt protein (CRP), quantiferon-Gold-testning för tuberkulos, snabb plasmareagin (RPR) för syfilis, angiotensinkonverterande enzym (ACE) och en bröströntgen för sarkoidos, antinukleär antikropp (ANA) och p-/c-ANCA. Också en historia av nyligen okulärt trauma eller intraokulär kirurgi måste noteras och föreslår sannolikt sympatisk oftalmi (SO) som den mer troliga diagnosen med tanke på den mycket liknande presentationen och patofysiologin som delas mellan SO och VKH .
för att stödja en diagnos av VKH i tvetydiga fall kan en ländryggspunktur utföras för att leta efter lymfocytisk och monocytisk pleocytos; detta används dock sällan kliniskt. Åttio procent av patienterna har pleocytos i cerebrospinalvätskan (CSF) inom en vecka och 97% har pleocytos inom tre veckor. Ökade nivåer av immunceller kan pågå i upp till åtta veckor efter sjukdomsuppkomsten . T-cellens ytmarkörprofiler är likartade mellan CSF och vattenhuman men skiljer sig från blodet. Detta föreslår CSF: s förmåga att exakt reflektera uveal inflammation vid VKH-sjukdom .
Tabell 1. Reviderade diagnostiska kriterier för Vogt-Koyanagi – Harada sjukdom
* från Tabell 1 i (15).
”fullständig Vogt-Koyanagi – Harada-sjukdom (kriterier 1 till 5 måste vara närvarande)
- ingen historia av penetrerande okulärt trauma eller kirurgi före uveit.
- inga kliniska eller laboratoriebevis som tyder på andra okulära sjukdomsenheter.
- bilateralt okulärt engagemang (a eller b måste uppfyllas, beroende på sjukdomsstadiet när patienten undersöks).
- tidiga manifestationer av sjukdom.
- Det måste finnas tecken på diffus koroidit (med eller utan främre uveit, glaskroppsinflammatorisk reaktion eller optisk diskhyperemi), som kan manifesteras som något av följande:
- tidiga manifestationer av sjukdom.
- fokala områden av subretinalvätska eller
- Bullous serösa retinala avdelningar.
- med tvetydiga fundusfynd; båda följande måste också vara närvarande:
- fokala områden med fördröjning i koroidal perfusion, multifokala områden med exakt läckage, stora placoidområden med hyperfluorescens, sammanslagning inom subretinalvätska och optisk nervfärgning (listad i ordning med sekventiellt utseende) genom fluoresceinangiografi och
- diffus koroidförtjockning, utan bevis på bakre sklerit genom ultraljud.
- sena manifestationer av sjukdom.
- historia som tyder på tidigare närvaro av fynd från 3a, och antingen både (2) och (3) nedan eller flera tecken från (3):
- okulär depigmentering (någon av följande manifestationer är tillräcklig): (a) Sunset glow fundus, eller (b) Sugiura-tecken.
- andra okulära tecken:
- Nummular chorioretinal depigmenterade ärr, eller
- Retinal pigmentepitel klumpning och/eller migration, eller
- återkommande eller kronisk främre uveit.
- Meningismus (sjukdomskänsla, feber, huvudvärk, illamående, buksmärtor, stelhet i nacke och rygg, eller en kombination av dessa faktorer; huvudvärk ensam är inte tillräckligt för att uppfylla definitionen av meningismus, Dock), eller
- Tinnitus, eller
- cerebrospinalvätska pleocytos.
- alopeci, eller
- polios, eller
- Vitiligo.
ofullständig Vogt-Koyanagi-Harada-sjukdom (kriterier 1 till 3 och antingen 4 eller 5 måste vara närvarande)
- ingen historia av penetrerande okulärt trauma eller kirurgi före uveit, och
- inga kliniska eller laboratoriebevis som tyder på andra okulära sjukdomsenheter och
- bilateralt okulärt engagemang.
- neurologiska / auditiva fynd; som definierat för fullständig Vogt-Koyanagi – Harada sjukdom ovan, eller
- integumentära fynd; som definierat för fullständig Vogt-Koyanagi-Harada sjukdom ovan.
sannolik Vogt-Koyanagi – Harada-sjukdom (isolerad okulär sjukdom; kriterier 1 till 3 måste vara närvarande)
- ingen historia av penetrerande okulärt trauma eller kirurgi före uveit.
- inga kliniska eller laboratoriebevis som tyder på andra okulära sjukdomsenheter.bilateralt okulärt engagemang enligt definitionen för fullständig Vogt-Koyanagi-Harada-sjukdom ovan. ”
testning / laboratoriearbete
i den första upparbetningen av VKH bör man överväga att erhålla följande tester:
- optisk koherens tomografi (OCT): I den akuta uveitiska fasen kommer OCT sannolikt att visa signifikant koroidförtjockning och serösa retinala avdelningar. De subretinala vätskeansamlingarna kan ha septationer som tros vara fibrinmembran och inflammatoriska produkter, vilket skapar en lobulär struktur som också kan ses på fluoresceinangiografi. I den konvalescenta fasen kan OCT upptäcka områden med retinal gallring efter löst inflammation efter kortikosteroidbehandling .
- B-scan ultraljud: I den akuta fasen kan ultraljud visa diffus bakre koroidal förtjockning, bakre skleral förtjockning, näthinneavskiljningar och glaskropp . Ciliär effusioner kan observeras med ultraljudsbiomikroskopi . Detta test är också användbart för att utesluta bakre sklerit.
- fluoresceinangiografi( FA): klassiskt avslöjar FA multifokala koroidala hypofluorescerande prickar i den tidiga fasen följt av flera fokala hyperfluorescerande områden med diffus läckage i den sena fasen . Färgämnet läcker genom RPE och ackumuleras i det subretinala utrymmet som omger de hyperfluorescerande prickarna. FA kan vara diagnostiskt användbart när VKH-sjukdomen presenterar utan extraokulära symtom. Optisk skiva hyperfluorescens och fönsterdefekter orsakade av atrofiska korioretinala ärr kan ses i mitten av periferin . FA i det kroniska återkommande stadiet av VKH-sjukdomen visar ospecifika fönsterfel på grund av RPE-skada, koroidal neovaskularisering och subretinal fibros .
- Indocyaningrön (ICG) angiografi: Tidig fas ICG visar hyperfluorescerande stromala kärl som indikerar koroidal vaskulopati och hypofluorescerande mörka prickar som motsvarar granulom och fördröjd fläckig fyllning av koroidal vaskulatur . Den sena fasen avslöjar fuzzy stromala vaskulära mönster och diffus koroidal hyperfluorescens. Skivhyperfluorescens tyder på allvarlig sjukdom. ICGA kan upptäcka subklinisk koroidal inflammation i mycket tidiga skeden eller till och med efter systemisk behandling .
- Lumbar punktering: pleocytos i cerebrospinalvätskan är närvarande hos majoriteten av VKH-patienterna. Lumbar punktering bör utföras tidigt i sjukdomsförloppet eftersom pleocytos kan lösa
behandling/hantering/riktlinjer
behandlingsmål i VKH inkluderar tidig diagnos och undertryckande av aktiv inflammation, tillsammans med förebyggande av återkommande inflammation och synhotande komplikationer, såsom glaukom, bullous retinal detachment och koroidal neovaskularisering.
systemisk kortikosteroidbehandling är den föredragna behandlingen för VKH-sjukdom, särskilt under det akuta uveitiska stadiet. Det har visats att administreringsvägen för kortikosteroid (oral kontra intravenös) inte påverkar synskärpan eller förekomsten av visuellt signifikanta komplikationer vid behandling av akut VKH . För allvarlig sjukdom är det föreslagna protokollet intravenös metylprednisolonadministration i tre dagar följt av oral högdos prednisonbehandling. Vid mild-måttlig sjukdom kan högdos oral prednison vara tillräcklig vid 1-2 mg/kg/dag. Steroiddosen ska långsamt avsmalna under cirka sex månader för att förhindra återfall . Aggressiv tidig behandling, tillsammans med seriell FA-testning som visar försvinnande av färgläckage genom RPE, kan bidra till att förhindra ytterligare sjukdomsprogression, återfall och extraokulära manifestationer . Topiska steroider och cykloplegika kan minska cellerna i den främre kammaren och glaskroppen.
intravitreala och sub-Tenoninjektioner av triamcinolon har använts för kortvarig kontroll av intraokulär inflammation under de akuta eller återkommande faserna; dessa lokala terapier bör övervägas vid motstridig sjukdom och hos patienter som dåligt tolererar de ogynnsamma systemiska biverkningarna av steroider med tanke på den utökade steroidkondensen. Intravitreal anti-VEGF-injektioner används ibland för kontroll av koroidal neovaskularisering och i fall av ihållande foveal serösa retinala avdelningar .
Steroidsparande medel inklusive antimetaboliter, kalcineurinhämmare, biologiska läkemedel, TNF-alfa-hämmare eller cytotoxiska medel kan användas för att behandla VKH och bör övervakas noggrant, ofta i samordning med en reumatologitjänst . Det har diskuterats om användningen av icke-steroida immunsuppressiva medel som förstahandsbehandling för VKH-sjukdom. En ny studie visade emellertid inga skillnader i resultat mellan tidig första linjens immunmodulerande behandling (IMT) och prednisonbehandling ensam . Vidare är immunsuppressiva och biologiska terapier dyra och kräver noggrann utvärdering före behandling samt frekvent uppföljning med blodarbete för att bedöma för allvarliga biverkningar.
i det kronisk-återkommande skedet kan frekvent återkommande föreslå resistens mot kortikosteroidbehandling och föreslår behov av steroidsparande immunmodulerande behandling . Det föredragna medlet för steroidresistent återfall eller steroidintolerans är cyklosporin . Infliximab, rituximab, adalimumab och interferon alfa-2a är biologiska medel som också har använts för att behandla eldfast uveit vid VKH-sjukdom.
för att behandla den främre uveit som ofta förknippas med akut VKH, bör topiska steroider (t.ex. prednisolonacetat 1%) och topisk cyloplegi (t. ex. cyklopentolat 1% eller atropin 1%) förskrivas beroende på graden av inflammation i främre kammaren.
okulära komplikationer är vanligtvis associerade med VKH-sjukdom. Med tanke på de många stegen och olika presentationer där en patient kan presentera med VKH, kan behandlingen försenas i många fall. I svåra former av VKH och i återkommande kan intraokulär inflammation vara svår att kontrollera och kan leda till strukturella skador. Över 50% av patienterna utvecklar relaterade komplikationer, inklusive katarakt, sekundär glaukom, koroidala neovaskulära membran, subretinal fibros eller en kombination av dessa (Figur 6) .
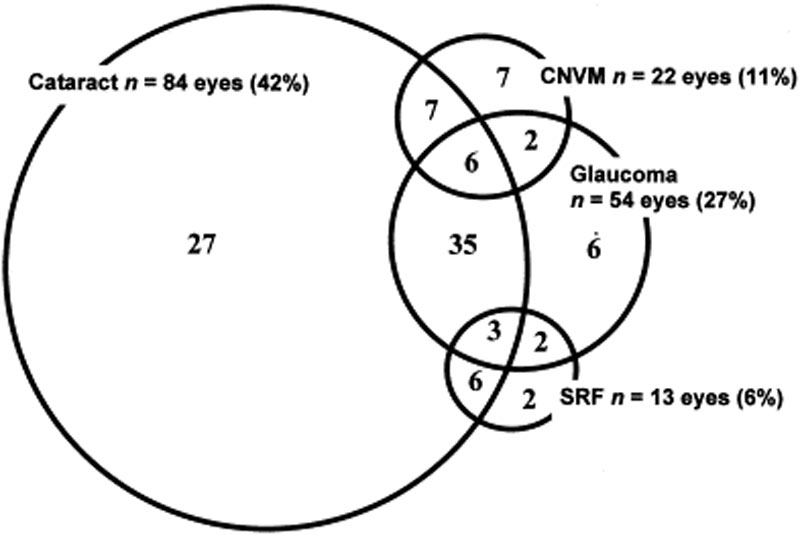
Figur 6: Venn-diagram som visar komplikationer hos VKH-patienter. (Används med tillstånd från Am J Ophthalmol. 2001;131(5):599-606 )
epidemiologi och etiologi
|
SIGNS
|
symptom
|
behandling/hantering
|
- Du L, Kijlstra a, Yang P. Vogt-Koyanagi-Harada sjukdom: nya insikter i patofysiologi, diagnos och behandling. Prog Retin Öga Res 2016; 52:84-111. https://PubMed.gov/26875727. DOI: 10.1016/j.preteyeres.2016.02.002
- Yamaki K, Gocho K, Hayakawa K, Kondo I, Sakuragi S. Tyrosinasfamiljproteiner är antigener specifika för Vogt-Koyanagi-Haradas sjukdom. J Immunol 2000; 165 (12):7323-7329. https://PubMed.gov/11120868
- Horie Y, Takemoto Y, Miyazaki A, Namba K, Kase S, Yoshida K, Ota M, Hasumi Y, Inoko H, Mizuki N, Ohno S. Tyrosinasgenfamilj och Vogt-Koyanagi – Harada sjukdom hos japanska patienter. Mol Vis 2006; 12: 1601-1605. https://PubMed.gov/17200659
- Ng JY, Luk FO, Lai TY, Pang CP. Påverkan av molekylär genetik i Vogt-Koyanagi-Harada sjukdom. J Oftalmisk Inflamm Infektera 2014; 4: 20. https://PubMed.gov/25097674. DOI: 10.1186 / s12348-014-0020-1
- Bowling B. Uveitis. Kanskis kliniska oftalmologi New York, New York: Elsevier; 2016; kapitel 11; s. 395-465.
- Yeh PT YC, Yang CH, Lin CP. Nonrhegmatogen Näthinneavlossning. I: Han är en av de mest kända i världen. Ryans näthinna. New York: Elsevier; 2018; kapitel 99; s. 1828-1849.
- Goto h rk, Rao N. Vogt-Koyanagi – Harada sjukdom. I: Schachat AP SS, Hinton DR, Wilkinson CP, Wiedemann P, redaktör. Ryans näthinna. New York, New York: Elsevier; 2018; kapitel 78; S. 1505-1515.Riddington l, Hall AJ, Tait B, Nicholson I, Varney M. Vogt-Koyanagi – Harada syndrom hos patienter med vietnamesiskt ursprung. Aust N Z J Oftalmol 1996; 24 (2):147-149. https://PubMed.gov/9199747
- Sugita S, Takase H, Kawaguchi T, Taguchi C, Mochizuki M. Korsreaktion mellan tyrosinaspeptider och cytomegalovirusantigen av T-celler från patienter med Vogt-Koyanagi-Harada-sjukdom. Int Oftalmol 2007; 27 (2-3):87-95. https://PubMed.gov/17253112. DOI: 10.1007 / s10792-006-9020-jag är en av de mest kända. Inflammation. Retinal Atlas. New York, New York: Elsevier 2017; Kapitel 4; s. 279-398.
- Rao N. Vogt-Koyanagi-Harada sjukdom. I: J YMaD, redaktör. Oftalmologi. New York, New York: Elsevier; 2014; kapitel 7.17; s. 761-763.
- Rao NA, Xu S, teckensnitt RL. Sympatisk oftalmi. En immunohistokemisk studie av epitelioida och jätteceller. Oftalmologi 1985; 92 (12): 1660-1662. https://PubMed.gov/4088616
- Nussenblatt RB. Vogt-Koyanagi-Harada Syndrom. I: Whitcup RBNaSM, redaktör. Uveit: grundläggande och klinisk praxis. 4: e upplagan ed: Elsevier; 2010; Kapitel Kapitel 24.Läs RW, Holland GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L, Pivetti-Pezzi P, Tessler HH, Usui M. reviderade diagnostiska kriterier för Vogt-Koyanagi – Harada sjukdom: rapport från en internationell nomenklaturutskott. Am J Oftalmol 2001; 131 (5):647-652. https://PubMed.gov/11336942
- Chung H, GD Choi. Klinisk analys av uveit. Koreanska J Oftalmol 1989; 3 (1): 33-37. https://PubMed.gov/2795939. DOI: 10.3341 / kjo.1989.3.1.33
- Abu El-Asrar AM, Al-Kharashi AS, Aldibhi H, Al-Fraykh H, kangave D. Vogt-Koyanagi-Harada sjukdom hos barn. Öga (Lond) 2008;22(9):1124-1131. https://PubMed.gov/17479116. DOI: 10.1038 / sj.ögon.6702859
- Martin TD, Rathinam SR, Cunningham ET. Prevalens, kliniska egenskaper och orsaker till synförlust hos barn med Vogt-Koyanagi-Harada sjukdom i södra Indien. Näthinnan 2010; 30 (7):1113-1121. https://PubMed.gov/20168275. DOI: 10.1097 / IAE.0b013e3181c96a87
- Forster DJ, grön RL, Rao NA. Unilateral manifestation av Vogt-Koyanagi – Harada syndrom hos ett 7-årigt barn. Am J Oftalmol 1991; 111 (3):380-382. https://PubMed.gov/2000916
- Yamamoto Y, Fukushima A, Nishino K, Koura Y, Komatsu T, Ueno H. Vogt-koyanagi-Harada sjukdom med debut hos äldre patienter i åldern 68 till 89 år. Jpn J Oftalmol 2007; 51 (1):60-63. https://PubMed.gov/17295144. DOI: 10.1007 / s10384-006-0379-0
- Wang Y, Chan CC. Könsskillnader i vogt-koyanagi-Harada sjukdom och sympatisk oftalmi. J Oftalmol 2014; 2014: 157803. https://PubMed.gov/24734166. DOI: 10.1155/2014/157803
- Nakao K, Abematsu N, Mizushima Y, Sakamoto T. optisk skiva svullnad i Vogt-Koyanagi – Harada sjukdom. Investera Oftalmol Vis Sci 2012; 53 (4):1917-1922. https://PubMed.gov/22408010. DOI: 10.1167 / iovs.11-8984
- Rao NA, Gupta A, Dustin L, Chee SP, Okada AA, Khairallah M, Bodaghi B, Lehoang P, Accorinti M, Mochizuki M, Prabriputaloong T, Läs RW. Frekvens av särskiljande kliniska egenskaper vid Vogt-Koyanagi-Harada sjukdom. Ophthalmology 2010;117(3):591-599, 599.e591. https://PubMed.gov/20036008. DOI: 10.1016/j.ophtha.2009.08.030
- Veerappan M, Fleischman D, Ulrich JN, Stinnett SS, Jaffe GJ, Allingham RR. The Relationship of Vogt-Koyanagi-Harada Syndrome to Ocular Hypertension and Glaucoma. Ocul Immunol Inflamm 2017;25(6):748-752. https://PubMed.gov/27438521. DOI: 10.1080/09273948.2016.1189578
- Baltmr A, Lightman S, Tomkins-Netzer O. Vogt-Koyanagi-Harada syndrome – current perspectives. Clin Ophthalmol 2016;10:2345-2361. https://PubMed.gov/27932857. DOI: 10.2147/OPTH.S94866
- Kitaichi n, Matoba H, Ohno S. Den positiva rollen av ländryggspunktur vid diagnosen Vogt-Koyanagi – Harada sjukdom: lymfocytundergrupper i vattenhuman och cerebrospinalvätska. Int Oftalmol 2007; 27 (2-3):97-103. https://PubMed.gov/17211585. DOI: 10.1007 / s10792-006-9016-7
- Oshima Y, Harino S, Hara Y, Tano Y. Indocyaningröna angiografiska fynd i Vogt-Koyanagi-Harada sjukdom. Am J Oftalmol 1996; 122 (1):58-66. https://PubMed.gov/8659599
- Läs RW, Yu f, Accorinti M, Bodaghi B, Chee SP, Fardeau C, Goto H, Holland GN, Kawashima H, Kojima E, Lehoang P, Lemaitre C, Okada AA, Pivetti-Pezzi P, Secchi A, se RF, Tabbara KF, Usui M, Rao NA. Utvärdering av effekten på resultaten av administreringsvägen för kortikosteroider vid akut Vogt-Koyanagi-Harada-sjukdom. Am J Oftalmol 2006; 142 (1):119-124. https://PubMed.gov/16815259. DOI: 10.1016 / j. ajo.2006.02.049
- Rubsamen PE, Gass JD. Vogt-Koyanagi-Harada syndrom. Klinisk kurs, Terapi och långsiktigt visuellt resultat. Arch Ophthalmol 1991;109(5):682-687. https://PubMed.gov/2025171
- Urzua CA, Velasquez V, Sabat P, Berger O, Ramirez S, Goecke A, V Jacobs DH, Gatica H, Guerrero J. tidigare immunmodulerande behandling är förknippad med bättre visuella resultat i en delmängd av patienter med Vogt-Koyanagi-Haradas sjukdom. Acta Oftalmol 2015; 93 (6):e475-480. https://PubMed.gov/25565265. DOI: 10.1111/aos.12648
- Läs RW, Rechodouni A, Butani N, Johnston R, LaBree LD, Smith RE, Rao NA. Komplikationer och prognostiska faktorer i Vogt-Koyanagi-Harada sjukdom. Am J Oftalmol 2001; 131 (5):599-606. https://PubMed.gov/11336934
föreslagna Citeringsformat
Mai AP, Tran C, Wilson CW, Fox AR, Boldt HC. Vogt-Koyanagi – Harada (VKH) sjukdom. EyeRounds.org. April 1, 2019. Tillgänglig från http://EyeRounds.org/cases/284-vogt-koyanagi-harada.htm
Leave a Reply