Vogt-Koyanagi-Harada (VKH) boala
autori: Anthony P. Mai, BS; Charlene Tran, BS; Caroline W. Wilson, MD; Austin R. Fox, MD; H. Culver Boldt, MD
1 aprilie 2019
prezentare inițială
plângere șef
vedere încețoșată și dureri de cap
istoria bolii prezente
o femeie vietnameză în vârstă de 44 de ani a prezentat departamentului de urgență o istorie de 10 zile de vedere încețoșată progresivă la ambii ochi și o istorie de trei zile de dureri de cap severe. Pierderea vederii centrale nu s-a îmbunătățit cu o refracție a optometristului ei. Durerile de cap occipitale severe s-au agravat odată cu mișcarea și au fost asociate cu stare generală de rău generalizată, oboseală extremă, fotofobie ușoară și rupere. Acetaminofenul a atenuat parțial durerea. ea a călătorit recent în Vietnam, dar a negat că a întâlnit contacte bolnave acolo. Ea a negat claudicația maxilarului, febra sau modificările de greutate. Ea a negat erupții cutanate, modificări ale auzului, tinitus, amețeli, amorțeală sau furnicături. A negat că ar fi avut vreodată tuberculoză. Nu avea antecedente de probleme de vedere anterioare, afecțiuni autoimune sau cancer.
antecedente oculare
- antecedente de chirurgie cosmetică a pleoapelor (blefaroplastie bilaterală) cu trei ani înainte
- nu există antecedente de traume sau boli oculare
antecedente medicale
nici unul
medicamente
acetaminofen după cum este necesar
alergii
nu există alergii cunoscute la medicamente
istoric familial
fără antecedente de boli oculare sau boli autoimune
istoric social
ea a imigrat din Vietnam cu câțiva ani înainte de prezentare. Este căsătorită și are trei copii. Lucrează la un salon de unghii. Nu consumă produse din tutun, alcool sau substanțe ilicite. Călătorește în Vietnam la fiecare șase până la douăsprezece luni.
revizuirea sistemelor
negativ, cu excepția a ceea ce este detaliat în istoricul bolii actuale
examinarea oculară
acuitatea vizuală cu / fără corecție (Snellen)
- ochiul drept (OD): 20/300 (fără îmbunătățire cu pinhole)
- ochiul stâng (OS): 20/60-2+2 (Nici o îmbunătățire cu pinhole)
motilitate oculară / aliniere
mișcări extraoculare complete la ambii ochi (OU)
presiunea intraoculară (Pio): (Tonopen)
- OD: 12 mmHg
- OS: 14 mmHg
elevi
- OD: 4 mm în întuneric, 3 mm în lumină, fără defect pupilar aferent relativ (RAPD)
- OS: 4 mm în întuneric, 3 mm în lumină, fără RAPD
confruntare câmpuri vizuale: (numărați
- od: scotom Central
- os: defect inferotemporal total
extern
normal pe ambele părți
examen lampă cu fantă
- capace/gene: normal ou
- conjunctiva/sclera: clar și liniștit ou
- cornee: 1+ eroziuni epiteliale punctate, fără precipitate keratice OU
- camera anterioară: celulă de urmărire și flare și adâncime ou
- Iris: Arhitectură normală ou
- lentilă: clar OU
examinare a fundului dilatat (DFE)
- vitros: urme celule vitroase anterioare ou
- Disc:
- OD: Grad 3 edem disc, hiperemic
- os: edem disc 2-3 grad, hiperemic
- raport cupă-disc: 0,0 ou
- macula:
- od: 3+ edem macular cistoid (CME) și lichid subretinal (SRF) care se extinde de la disc la macula temporală. Fără lipide sau exudate. Choroid care apare în mlaștină.
- OS: 2 + CME și SRF care se extind de pe disc prin fovea. 1-2 + lipide liniare care se extind de la disc spre fovee. Choroid care apare în mlaștină.
- vase:
- OD: înveliș temporal
- OS: Normal
- periferie:
- OD: smoc retinian chistic anterior ecuatorului la 10: 30
- OS: SRF superficial anterior ecuatorului la 4:00
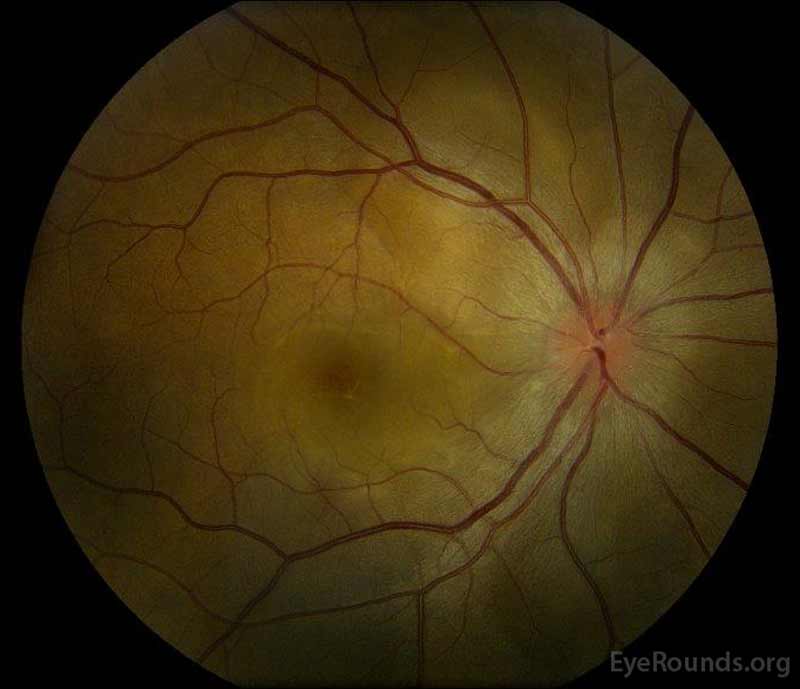 |
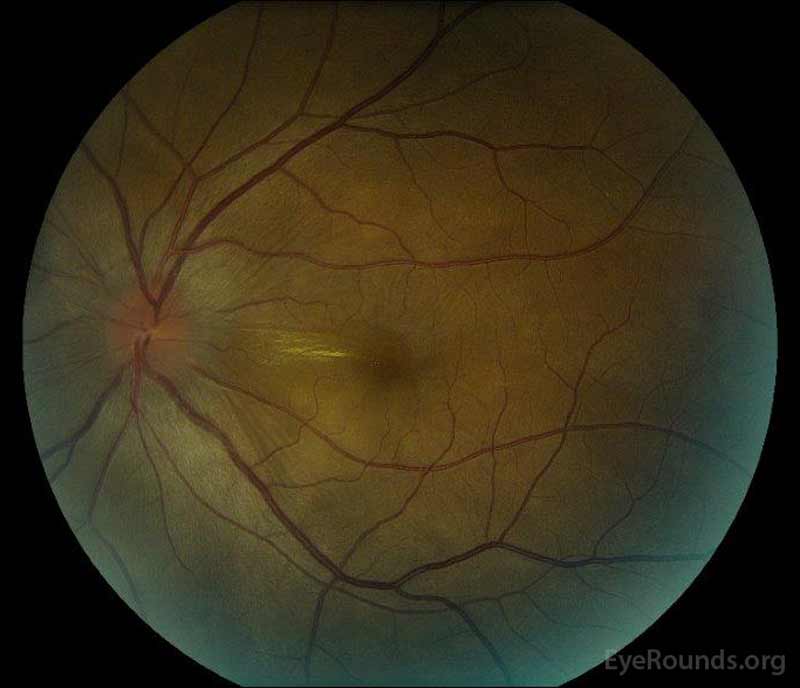 |
figura 1: fotografii de fond color la prezentare: (imaginea din stânga) ochiul drept prezintă edem de disc și hiperemie ușoară, precum și lichid subretinal care se extinde de pe disc temporal prin maculă. Există, de asemenea, un detașament focal seros de retină superotemporal pe disc, de-a lungul arcadei superioare. (Right image) The left eye has disc edema and mild hyperemia, along with subretinal fluid extending from the disc to the macula and linear lipid deposits in the nasal macula.
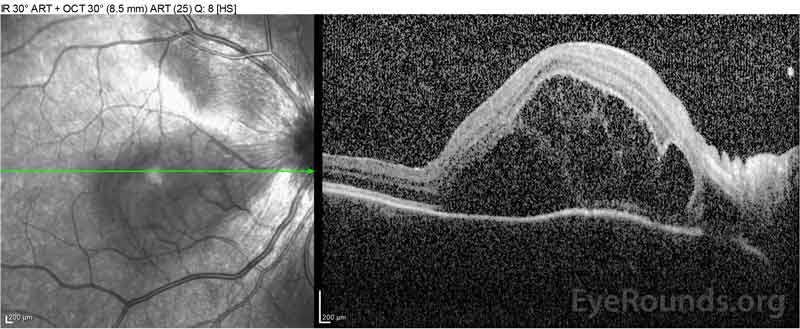 |
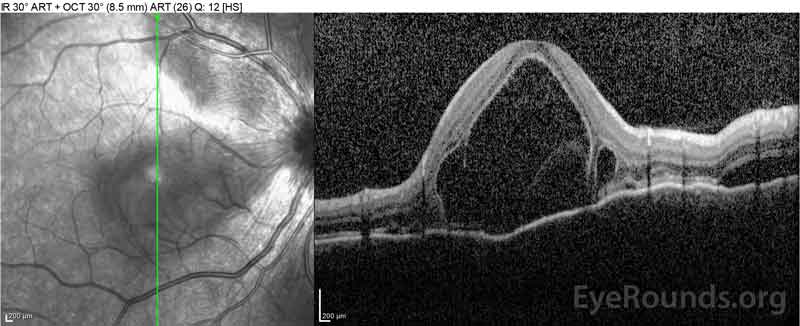 |
 |
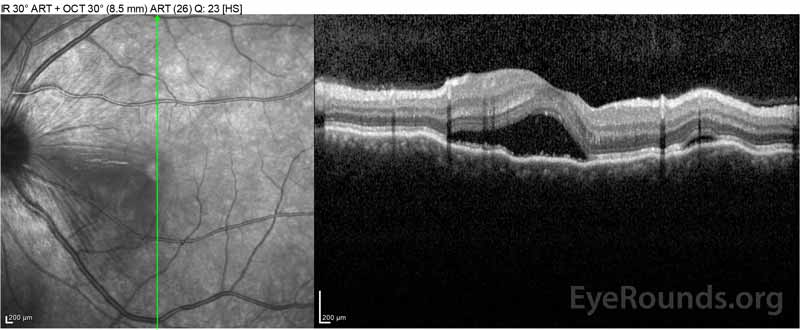 |
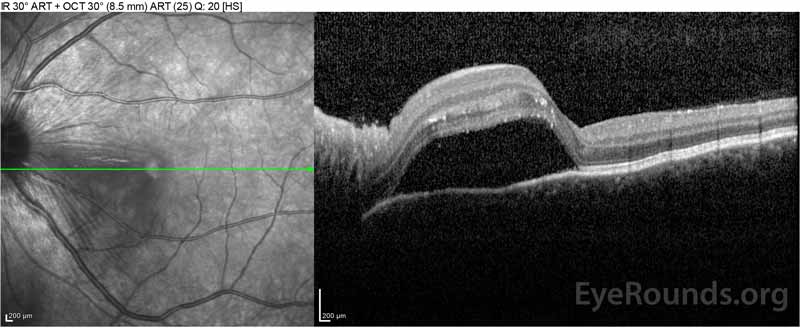
Figure 2: Tomografia de coerență optică (OCT) a ochiului drept (panourile superioare) prezintă o detașare seroasă a retinei care implică foveea cu lichid intraretinal extins, întreruperea straturilor retiniene exterioare și ondulațiile coroidului îngroșat. OCT a ochiului stâng (panourile inferioare) prezintă o detașare seroasă a retinei în macula nazală care se extinde până la fovee.
 |
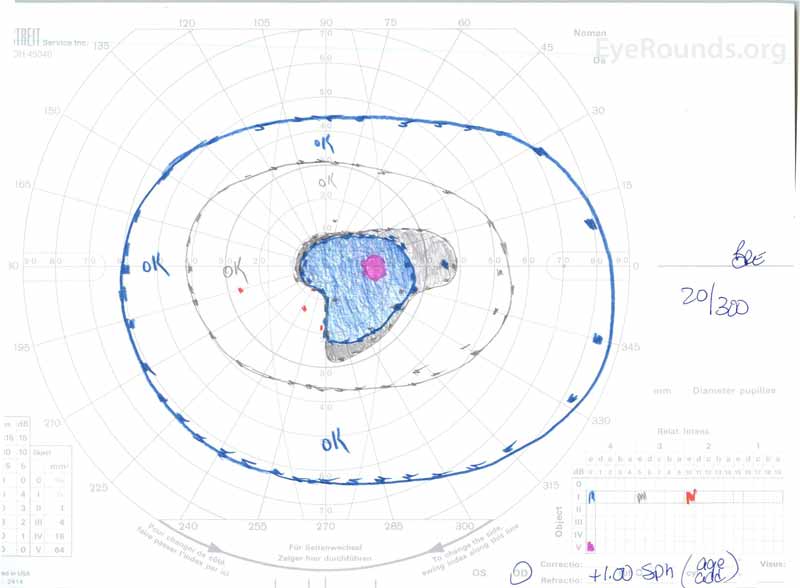 |
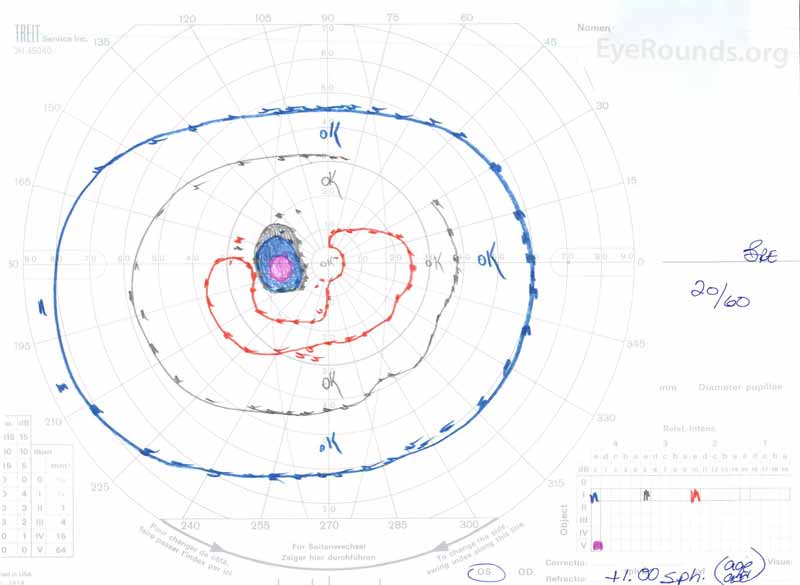
figura 3: Câmpuri vizuale Goldman( GVF), OU. (Imaginea din stânga) OS arată un punct orb fiziologic mărit și un scotom Central ușor. (Imaginea din dreapta) OD prezintă un scotom central moderat dens care încorporează punctul orb fiziologic și se extinde inferotemporal.
B-scan: nu există semne de sclerită, opacități vitreale ușoare/celule inferioare
diagnostic diferențial
- epiteliopatie pigmentară placoidă multifocală posterioară acută (APMPPE)
- corioretinopatie seroasă centrală
- nevrită optică
- Panuveită
- boală autoimună (de exemplu, les, sarcoidoză)
infecție (e.(de exemplu, limfom ocular)
lucru-UP
hemogramă completă
numărul de celule albe din sânge: 4,9 k/mm3 (ref: 3,7-10,5)
numărul de celule roșii din sânge 3,99 m/mm3 (ref: 4,0-5,2)
hemoglobină 11,6 g/dl (Ref: 11,9-15,5)
hematocrit 35 % (ref: 35-47)
Basic metabolic panel
Sodium 138 mEq/L (Ref: 135-145)
Potassium 4.3 mEq/L (Ref: 3.5-5.0)
Chloride 107 mEq/L (Ref: 95-107)
CO2 20 mEq/L (Ref: 22-29)
Blood urea nitrogen 16 mEq/dL (Ref: 10-20)
Creatinine 0.7 mg/dL (Ref: 0.5-1.0)
C-reactive Protein (CRP): <0.5 mg/dL (Ref: <=0.5)
Erythrocyte sedimentation rate (ESR): 12 mm/Hr (Ref: 0-20)
Angiotensin–converting enzyme (ACE): 13 U/L (Ref: 8-52)
QuantiFERON-TB aur: negativ
fier, sânge 54 micrograme/dL (Ref: 37-145)
capacitate totală de legare a fierului 379 micrograme/dL (Ref: 250-425)
curs clinic
pacientul a fost evaluat inițial de Departamentul de urgență, având în vedere plângerile sale de dureri de cap severe cu debut nou și pierderea vederii. Tomografia computerizată a creierului (CT) și imagistica prin rezonanță magnetică (RMN) au fost de neimaginat. ESR și CRP au fost la niveluri normale. Clinica de Oftalmologie a evaluat-o a doua zi și a găsit detașamente bilaterale seroase de retină și panuveită. ACE și QuantiFERON-TB Gold labs au fost ambele negative. A fost diagnosticată cu boala Vogt-Koyanagi-Harada pe baza prezentării sale clinice și a descendenței asiatice. Ea a fost tratată cu 80 mg de prednison zilnic, acetaminofen după cum este necesar pentru durerile de cap și suplimentarea cu vitamina D și calciu. Durerile de cap s-au rezolvat rapid, iar acuitatea vizuală s-a îmbunătățit constant în următoarele două săptămâni. Doza de prednison a fost apoi redusă la 40 mg pe parcursul a trei săptămâni, cu rezolvarea continuă a simptomelor și îmbunătățirea acuității vizuale. Ea nu a avut nici o recurență de dureri de cap sau agravarea vederii în timpul conic prednison. La cea mai recentă întâlnire, ea a redus la 5 mg în fiecare zi, fără revenirea simptomelor. Acuitatea vizuală la acea vizită de urmărire a fost de 20/15-2 OD și 20/20+2 OS, iar OCT macular a prezentat o rezoluție completă a edemului discului și a detașărilor seroase ale retinei la ambii ochi (figura 4).
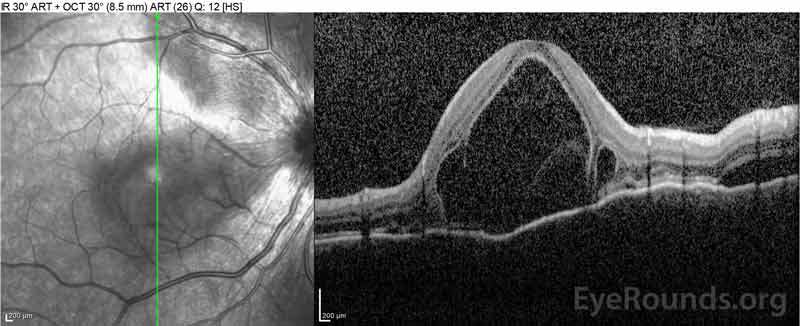
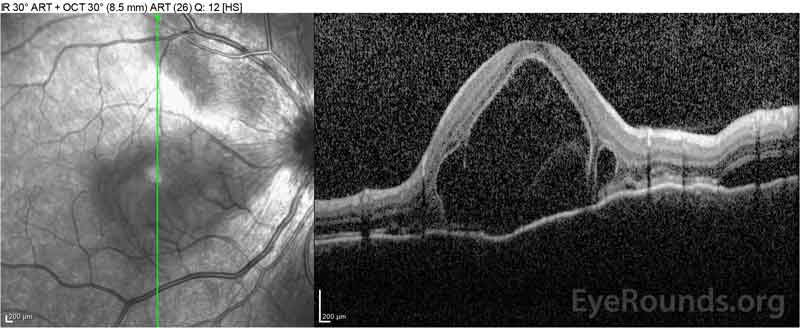
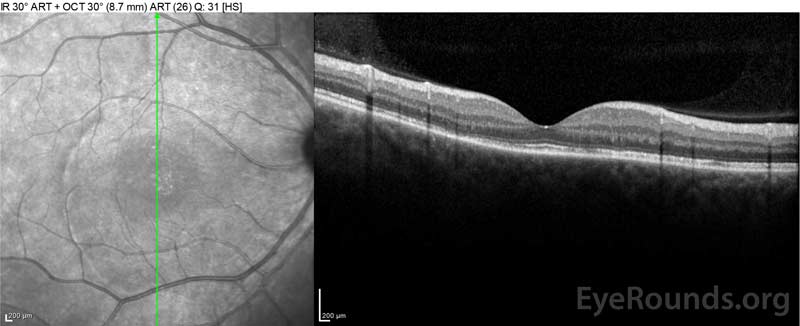
Figura 4: Tomografie de coerență optică care prezintă lichid subretinal la momentul inițial (sus) și cursul rezoluției la o săptămână (mijloc) și cinci săptămâni (jos) în timp ce se administrează o conicitate orală de prednison cu doze mari. Rețineți netezirea ondulațiilor coroidiene cu tratament.
 |
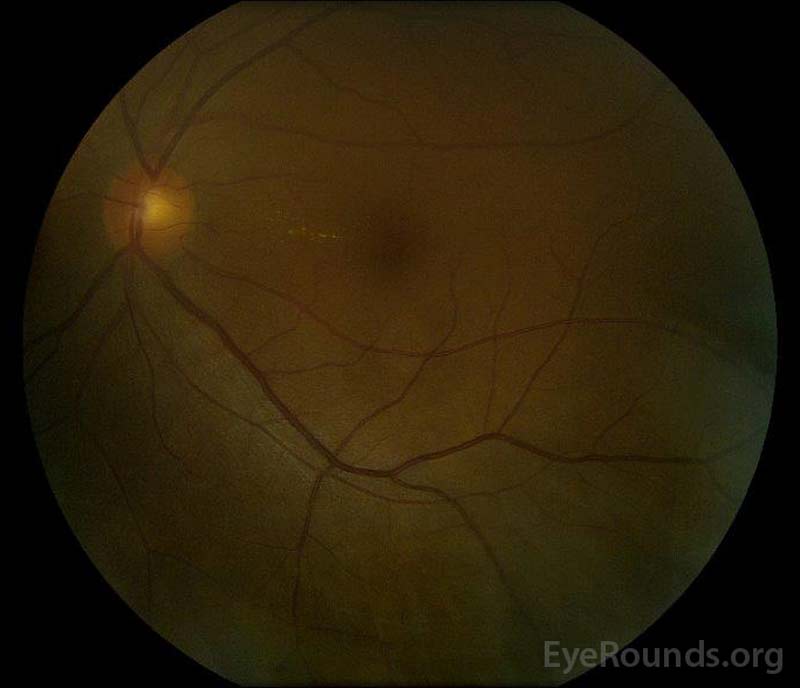 |
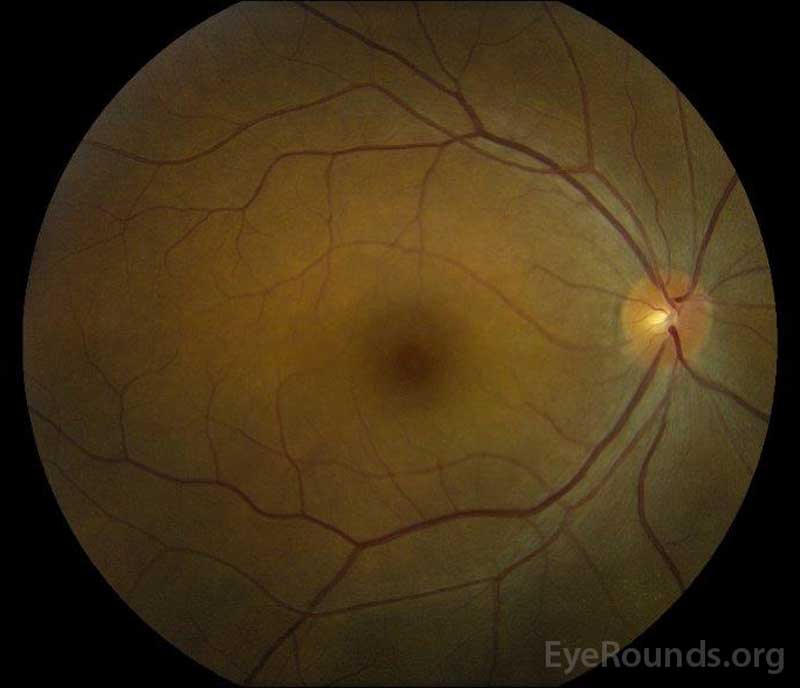
figura 5: Fotografia fundului de culoare a ochilor drepți (a) și stângi (B) în timpul fazei de convalescență care demonstrează îmbunătățirea fluidului subretinal și a edemului discului.
diagnostic
incomplet boala Vogt-Koyanagi-Harada
discuție
boala Vogt-Koyanagi-Harada (VKH) este o afecțiune autoimună sistemică caracterizată prin panuveită granulomatoasă bilaterală non-necrotizantă asociată cu modificări integumentare extraoculare, cum ar fi polioza și vitiligo, și inflamația care afectează uvea, urechea internă, părul și meningele. Boala Harada este uveita izolată fără semnele sau simptomele sistemice ale VKH.
etiologie
etiologia bolii VKH este încă în mare parte necunoscută, în ciuda eforturilor actuale de cercetare. Se crede că este o boală autoimună dobândită care implică hipersensibilitate mediată de celulele T la auto-antigene melanocitare, cu o predispoziție genetică subiacentă și posibil declanșator microbian . Tirozinaza și peptidele legate de tirozinază sunt antigene melanocitare care au fost sugerate ca ținte ale proceselor autoimune în VKH . Cu toate acestea, riscul crescut de boală VKH nu a fost asociat cu familia genei tirozinazei, potrivit unui studiu .
Din cauza prevalenței crescute în rândul anumitor grupuri etnice și de gen, se crede că există o predispoziție genetică în patogeneza VKH. Gene Multiple, inclusiv antigenul leucocitar uman (HLA) și genele interleukinei (IL), au fost asociate cu VKH în diferite populații etnice . Receptorii HLA sunt complexe majore de histocompatibilitate la om care prezintă peptide sistemului imunitar. HLA-DR1, HLA-DR4, HLA-DRB1*0405 și HLA-DRw53 sunt mai multe haplotipuri găsite la pacienții cu boală VKH . HLA-DR4 este mai frecventă la japonezi și hispanici, în timp ce HLA-DRB1*0405 este mai frecventă la pacienții coreeni și din Orientul Mijlociu . Atât alelele HLA-DR4, cât și HLA-DRB1*0405 se găsesc la pacienții Vietnamezi . Receptorul HLA-DRB1 se leagă de antigenele melanocitelor în diferite capacități. În ciuda acestor asociații, testarea genetică nu este recomandată în acest moment.
având în vedere simptomele prodromale obișnuite care însoțesc VKH, inclusiv febră, dureri de cap, meningism și tinitus, a fost sugerată o etiologie virală incitantă ca declanșator al debutului VKH prin mecanisme de mimetism molecular la pacienții predispuși genetic. Învelișul citomegalovirusului glicoproteina H are o omologie semnificativă a aminoacizilor cu peptida tirozinazei, iar infecția cu CMV poate declanșa VKH prin mimetism molecular (adică recunoașterea de către receptorii HLA-clasa II) . Virusul Ebstein-bar (EBV) a fost, de asemenea, implicat. Cu toate acestea, nu au existat dovezi definitive cu privire la o etiologie virală a VKH și rămâne neclar ce declanșează răspunsul autoimun VKH .
Fiziopatologie
există patru faze clasice ale VKH care pot avea prezentări variabile: prodromal, uveitic acut, convalescent și cronic-recurent. Modificările histopatologice încep de obicei în faza acută .
faza uveitică acută se caracterizează prin îngroșarea uveală bilaterală secundară inflamației granulomatoase. Granuloamele constau din limfocite, macrofage și celule epiteliale și gigantice umplute cu granule . Deși celulele epitelioide au fost considerate anterior a fi melanocite modificate, un studiu imunohistochimic de urmărire a sugerat o origine din macrofage tisulare . Granuloamele umplute cu histiocite epitelioide, denumite noduli Dalen-Fuchs, pot fi adesea observate între epiteliul pigmentar retinian (RPE) și membrana lui Bruch. Inflamația granulomatoasă uveală duce la îngroșarea coroidală și detașamente retiniene exudative umplute cu lichid proteic. În plus, pleocitoza (i.E., creșterea numărului de celule) poate fi prezentă în camera anterioară și vitroasă .
faza convalescentă este identificată prin depigmentarea zonelor coroide și extraoculare, inclusiv a pielii și părului periocular. Un coroid depigmentat setat pe un nerv optic palid dă impresia unui fundus „sunset-glow”, care este o caracteristică clasică a acestei faze a VKH . În plus, nodulii Dalen-Fuchs devin mai proeminenți sub RPE în faza convalescentă .
faza cronică-recurentă se caracterizează prin scăderea grosimii coroidiene, rezoluția detașamentelor retinale seroase, vitrita cronică ușoară și inflamația recurentă a segmentului anterior granulomatos. Neovascularizarea coroidiană (CNV) și fibroza subretinală se pot dezvolta în această fază și sunt indicatori ai progresiei severe a bolii . Cataracta și glaucomul secundar sunt alte complicații ale inflamației de lungă durată sau recurente în această fază .
Epidemiologie
VKH este răspândită în rasele cu pigment de piele mai închis, în special asiatici, Sud-Americani, Orientul Mijlociu și nativi americani. Boala VKH reprezintă >10% din uveita la aceste populații . Doar 1-4% din cazurile de uveită sunt considerate a fi secundare bolii VKH în Statele Unite (7). În Statele Unite, s-a constatat că majoritatea cazurilor de VKH afectează indivizii asiatici, hispanici și/sau nativi americani decenți . Interesant este că boala VKH afectează rar africanii, în ciuda pigmentării lor întunecate . Incidența bolii VKH variază foarte mult între subgrupurile rasiale din țările vecine . De exemplu, incidența VKH în Coreea este de numai 2%, mult mai mică decât cea găsită în Japonia și China . VKH are un debut tipic de 20 până la 50 de ani ; cu toate acestea, studiile sugerează că 3,1-13,4% din cazurile VKH sunt pacienți pediatri și 10% din cazuri sunt în vârstă de 65 de ani . În mod clasic, se crede că VKH are o predilecție pentru sexul feminin și, deși majoritatea studiilor arată că VKH afectează în mod disproporționat femeile, câteva studii au arătat o predispoziție masculină sau nicio predispoziție de gen .
semne/simptome
după cum sa menționat mai sus, cele patru etape ale bolii VKH sunt prodromale, uveitice, convalescente și recurente cronice. Fiecare etapă prezintă caracteristici clinice distincte.
- Prodromal: această etapă inițială se poate prezenta ca o boală asemănătoare gripei cu simptome predominant constituționale, cum ar fi dureri de cap, amețeli, febră, oboseală și / sau greață. Au fost raportate simptome neurologice ale meningitei, paliilor nervilor cranieni și nevritei optice, precum și simptome auditive ale tinitusului, disacuziei și vertijului . Fotofobia, vederea încețoșată, flocoanele și/sau durerile oculare încep de obicei în 48 de ore de la simptomele prodromale . Faza prodromală durează de obicei de la câteva zile până la săptămâni.
- Uveitic acut: această etapă include vedere încețoșată, fotofobie, injecție conjunctivală și dureri oculare. Poate exista uveită anterioară ușoară care la început apare non-granulomatoasă. Debutul Unilateral trece de obicei la implicarea bilaterală în decurs de 1-2 săptămâni. Se poate dezvolta uveita anterioară granulomatoasă cu precipitații keratice de grăsime de oaie. Rezultatele examenului Posterior pot include edemul nervului optic și hiperemia , zonele multifocale ale coroiditei, mai multe zone de detașamente retiniene seroase localizate la fundul posterior, îngroșarea coroidală, pliurile corioretinale radiante și vitrita . Detașamentele retiniene seroase pot forma un model de trifoi în fundul posterior și ar putea progresa către detașamente buloase extinse în cazuri severe . Glaucomul inflamator acut a fost asociat cu această fază a bolii și se poate prezenta cu o cameră anterioară superficială secundară edemului corpului ciliar, imitând închiderea cu unghi acut . Durata fazei uveitice acute depinde de diagnosticul și managementul prompt.
- Uveitic cronic sau Convalescent: această etapă se dezvoltă de obicei la câteva săptămâni după faza acută și se caracterizează prin vitiligo (de exemplu, față, mâini, umeri sau spate), polioză și alopecie . Depigmentarea în apropierea limbului cornean, cunoscută sub numele de semnul lui Sugiura, poate fi văzută la o lună după debutul bolii ; cu toate acestea, acest semn este rar văzut în afara populației japoneze . Depigmentarea coroidală apare de obicei în câteva luni și are ca rezultat culoarea roșu-portocaliu strălucitor a coroidului și clasicul „fundus sunset glow.”Sunset glow fundus este considerat a fi cel mai important și predictiv în diagnosticul VKH cronic . Cicatricile corioretinale bine definite, rotunde, nummulare se pot forma în periferia mijlocie. Faza uveitică cronică durează de obicei câteva luni.
- cronică-recurentă: Această etapă se caracterizează prin episoade recurente de uveită anterioară granulomatoasă cu precipitate cheratice de grăsime de oaie, noduli de iris, depigmentare a irisului, sinechii posterioare, cataractă subcapsulară posterioară, glaucom secundar, membrane neovasculare coroidiene și, în cele din urmă, fibroza subretinală și atrofia corioretinală nummulară . Faza cronică se dezvoltă de obicei la cel puțin șase luni de la prezentarea inițială. Detașările retiniene seroase prezente în timpul fazelor acute și convalescente nu se repetă de obicei cu tratamentul agresiv cu corticosteroizi .
criterii de Diagnostic
cele mai recente criterii de diagnostic, denumite criteriile de Diagnostic revizuite (RDC) pentru VKH, au fost definite în 1999 la primul Atelier internațional privind VKH . Acestea sunt prezentate în tabelul 1. RDC sunt utile prin faptul că împart VKH în trei categorii diferite de diagnostic bazate pe faza bolii în care un pacient prezintă: complet, incomplet și probabil. Această clasificare a bolii permite o gestionare adecvată și timpurie a bolii ” probabile „care poate ajuta la prevenirea progresiei către boala” completă”.
elaborarea altor cauze ale inflamației oculare, atât infecțioase, cât și auto-inflamatorii, sunt esențiale. Acestea pot include rata de sedimentare a eritrocitelor (ESR), proteina C reactivă (CRP), testarea cuantiferon-aur pentru tuberculoză, reagina plasmatică rapidă (RPR) pentru sifilis, enzima de conversie a angiotensinei (ACE) și o radiografie toracică pentru sarcoidoză, anticorp antinuclear (ANA) și p-/c-ANCA. De asemenea, trebuie remarcat un istoric al traumatismelor oculare recente sau al chirurgiei intraoculare și sugerează probabil oftalmia simpatică (SO) ca diagnostic mai probabil, având în vedere prezentarea și fiziopatologia foarte asemănătoare între SO și VKH .
pentru a susține un diagnostic de VKH în cazuri echivoce, se poate efectua o puncție lombară pentru a căuta pleocitoză limfocitară și monocitară; cu toate acestea, aceasta este rareori utilizată clinic. Optzeci la sută dintre pacienți au pleocitoză în lichidul cefalorahidian (LCR) în decurs de o săptămână și 97% au pleocitoză în decurs de trei săptămâni. Nivelurile crescute de celule imune pot dura până la opt săptămâni după debutul bolii . Profilele markerului suprafeței celulelor T sunt similare între LCR și umoarea apoasă, dar diferite de sânge. Acest lucru sugerează capacitatea LCR de a reflecta cu exactitate inflamația uveală în boala VKH .
Tabelul 1. Criterii de Diagnostic revizuite pentru boala Vogt-Koyanagi-Harada
*din tabelul 1 din (15).
„boala Vogt-Koyanagi-Harada completă (criteriile de la 1 la 5 trebuie să fie prezente)
- nu există antecedente de traumatisme oculare penetrante sau intervenții chirurgicale anterioare debutului inițial al uveitei.
- nu există dovezi clinice sau de laborator care să sugereze alte entități ale bolii oculare.
- implicarea oculară bilaterală (a sau b trebuie îndeplinite, în funcție de stadiul bolii la examinarea pacientului).
- manifestări timpurii ale bolii.
- trebuie să existe dovezi ale unei coroidite difuze (cu sau fără uveită anterioară, reacție inflamatorie vitroasă sau hiperemie a discului optic), care se poate manifesta ca una dintre următoarele:
- manifestări timpurii ale bolii.
- zone focale ale lichidului subretinal sau
- detașamente retiniene seroase buloase.
- cu constatări echivoce ale fundului; trebuie să fie prezente și următoarele:
- zone focale de întârziere a perfuziei coroidiene, zone multifocale de scurgere punctuală, zone placoide mari de hiperfluorescență, gruparea în lichidul subretinal și colorarea nervului optic (enumerate în ordinea aspectului secvențial) prin angiografie cu fluoresceină și
- îngroșare coroidală difuză, fără dovezi de sclerită posterioară prin ultrasonografie.
- manifestări tardive ale bolii.
- istoric sugestiv pentru prezența anterioară a constatărilor de la 3a și fie ambele (2) și (3) sub sau semne multiple de la (3):
- depigmentare oculară (oricare dintre următoarele manifestări este suficientă): (a) Sunset glow fundus sau (b) Sugiura sign.
- alte semne oculare:
- cicatrici depigmentate corioretinale nummulare sau
- aglomerarea epiteliului pigmentar retinian și/sau migrarea sau
- uveită anterioară recurentă sau cronică.
- meningism (stare generală de rău, febră, cefalee, greață, dureri abdominale, rigiditate a gâtului și a spatelui sau o combinație a acestor factori; durerea de cap singură nu este suficientă pentru a satisface definiția meningismului) sau
- tinitus sau
- pleocitoza lichidului cefalorahidian.
- Alopecia sau
- polioza sau
- Vitiligo.
boala Vogt-Koyanagi-Harada incompletă (criteriile 1-3 și 4 sau 5 trebuie să fie prezente)
- nu există antecedente de traumatisme oculare penetrante sau intervenții chirurgicale anterioare apariției inițiale a uveitei și
- nu există dovezi clinice sau de laborator care să sugereze alte entități ale bolii oculare și
- implicare oculară bilaterală.
- constatări neurologice/auditive; așa cum sunt definite pentru boala Vogt-Koyanagi-Harada completă de mai sus sau
- constatări tegumentare; așa cum sunt definite pentru boala Vogt-Koyanagi-Harada completă de mai sus.
probabil boala Vogt-Koyanagi-Harada (boală oculară izolată; criteriile de la 1 la 3 trebuie să fie prezente)
- nu există antecedente de traumatisme oculare penetrante sau intervenții chirurgicale anterioare debutului inițial al uveitei.
- nu există dovezi clinice sau de laborator care să sugereze alte entități ale bolii oculare.
- implicarea oculară bilaterală, așa cum este definită pentru boala Vogt-Koyanagi-Harada completă de mai sus. „
testare / laborator de lucru-up
în workup inițială a VKH, ar trebui să ia în considerare obținerea următoarele teste:
- tomografie coerență optică (OCT): În faza uveitică acută, OCT va prezenta probabil o îngroșare coroidală semnificativă și detașamente retiniene seroase. Acumulările de lichid subretinal pot avea septări considerate a fi membrane de fibrină și produse inflamatorii, creând o structură lobulară care poate fi văzută și pe angiografia cu fluoresceină. În faza de convalescență, OCT poate detecta zone de subțiere a retinei în urma inflamației rezolvate după tratamentul cu corticosteroizi .
- B-scan ultrasonografie: În faza acută, ultrasonografia poate prezenta îngroșare coroidală posterioară difuză, îngroșare sclerală posterioară, detașamente retiniene și opacități vitroase . Efuziunile ciliare pot fi observate cu biomicroscopie cu ultrasunete . Acest test este util și pentru excluderea scleritei posterioare.
- angiografia fluoresceinei( FA): clasic, FA dezvăluie puncte hipofluorescente coroidale multifocale în faza timpurie urmată de multiple zone hiperfluorescente focale cu scurgeri difuze în faza târzie . Colorantul se scurge prin RPE și se acumulează în spațiul subretinal care înconjoară punctele hiperfluorescente. FA poate fi util din punct de vedere diagnostic atunci când boala VKH se prezintă fără simptome extraoculare. Hiperfluorescența discului Optic și defectele de fereastră cauzate de cicatricile corioretinale atrofice pot fi observate la periferia mijlocie . FA în stadiul cronic-recurent al bolii VKH prezintă defecte nespecifice ale ferestrei datorate deteriorării RPE, neovascularizării coroidiene și fibrozei subretinale .
- indocianină verde (ICG) angiografie: Faza timpurie ICG descrie vasele stromale hiperfluorescente care indică vasculopatia coroidiană și punctele întunecate hipofluorescente care corespund granuloamelor și umplerii neuniforme întârziate a vasculaturii coroidiene . Faza târzie relevă modele vasculare stromale fuzzy și hiperfluorescență coroidiană difuză. Hiperfluorescența discului sugerează o boală severă. ICGA poate detecta inflamația coroidiană subclinică în stadii foarte timpurii sau chiar după terapia sistemică .
- puncția lombară: pleocitoza în lichidul cefalorahidian este prezentă la majoritatea pacienților cu VKH. Puncția lombară trebuie efectuată la începutul cursului bolii, deoarece pleocitoza poate rezolva
tratament/Management/orientări
obiectivele tratamentului în VKH includ diagnosticul precoce și suprimarea inflamației active, împreună cu prevenirea inflamației recurente și a complicațiilor care pun în pericol vederea, cum ar fi glaucomul, detașarea retinei buloase și neovascularizarea coroidiană.
tratamentul sistemic cu corticosteroizi este terapia preferată pentru boala VKH, în special în stadiul uveitic acut. S-a demonstrat că calea de administrare a corticosteroizilor (oral versus intravenos) nu afectează acuitatea vizuală sau apariția complicațiilor semnificative vizual în tratamentul VKH acut . Pentru boala severă, protocolul sugerat este administrarea intravenoasă de metilprednisolon timp de trei zile, urmată de tratament oral cu doze mari de prednison. În boala ușoară-moderată, prednisonul oral cu doze mari poate fi suficient la 1-2mg/kg/zi. Doza de steroizi trebuie să fie încet conic peste aproximativ șase luni pentru a preveni recurența . Tratamentul precoce agresiv, alături de testarea fa în serie care arată dispariția scurgerilor de colorant prin RPE, poate ajuta la prevenirea progresiei ulterioare a bolii, recurenței și manifestărilor extraoculare . Steroizii topici și cicloplegicele pot scădea celulele din camera anterioară și umorul vitros.
injecțiile intravitroase și sub-Tenon de triamcinolonă au fost utilizate pentru controlul pe termen scurt al inflamației intraoculare în timpul fazelor acute sau recurente; aceste terapii locale trebuie luate în considerare în cazul bolii recalcitrante și la pacienții care tolerează slab efectele secundare sistemice nefavorabile ale steroizilor, având în vedere conicitatea extinsă a steroizilor. Injecțiile intravitroase anti-VEGF sunt uneori utilizate pentru controlul neovascularizării coroidiene și în cazurile de detașamente retiniene seroase foveale persistente .
agenții care economisesc steroizi, inclusiv antimetaboliți, inhibitori de calcineurină, biologici, inhibitori TNF-alfa sau agenți citotoxici pot fi utilizați pentru a trata VKH și trebuie monitorizați cu atenție, adesea în coordonare cu un serviciu de Reumatologie . Au existat discuții în curs cu privire la utilizarea medicamentelor imunosupresoare nesteroidiene ca terapie de primă linie pentru boala VKH. Cu toate acestea, un studiu recent nu a evidențiat diferențe în rezultatele dintre tratamentul imunomodulator de primă linie (IMT) și tratamentul cu prednison în monoterapie . În plus, terapiile imunosupresoare și biologice sunt costisitoare și necesită o evaluare atentă înainte de tratament, precum și o urmărire frecventă cu analize de sânge pentru a evalua efectele secundare grave.
în stadiul cronic-recurent, recurența frecventă poate sugera rezistență la terapia cu corticosteroizi și sugerează necesitatea unui tratament imunomodulator care economisește steroizi . Agentul preferat pentru recurența rezistentă la steroizi sau intoleranța la steroizi este ciclosporina . Infliximab, rituximab, adalimumab și interferon alfa-2a sunt agenți biologici care au fost, de asemenea, utilizați pentru a trata uveita refractară în boala VKH.
pentru a trata uveita anterioară asociată adesea cu VKH acut, trebuie prescrise steroizi topici (de exemplu, acetat de prednisolon 1%) și cyloplegia topică (de exemplu, ciclopentolat 1% sau atropină 1%) în funcție de gradul de inflamație a camerei anterioare.
complicațiile oculare sunt frecvent asociate cu boala VKH. Având în vedere multiplele etape și varietatea prezentărilor în care un pacient se poate prezenta cu VKH, tratamentul poate fi întârziat în multe cazuri. În formele severe de VKH și în recurențe, inflamația intraoculară poate fi dificil de controlat și poate duce la leziuni structurale. Peste 50% dintre pacienți dezvoltă complicații asociate, inclusiv cataractă, glaucom secundar, membrane neovasculare coroidiene, fibroză subretinală sau o combinație a acestora (Figura 6) .
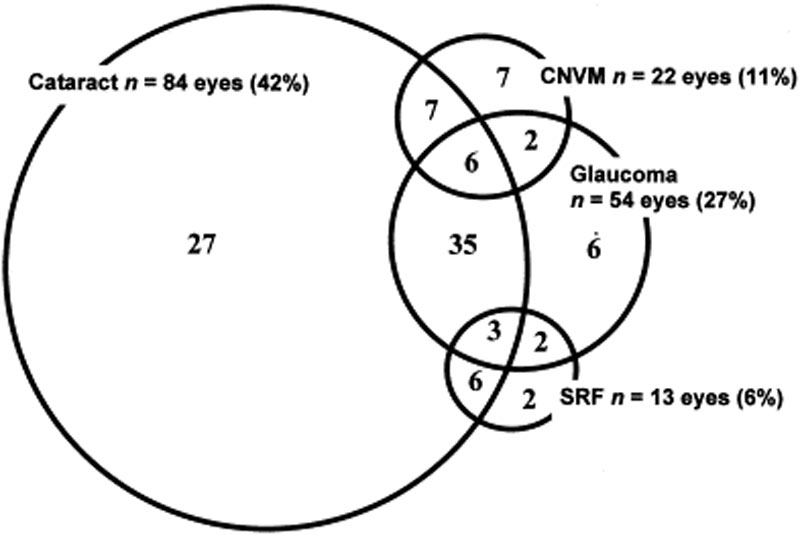
Figura 6: Diagrama Venn care prezintă complicații la pacienții cu VKH. (Folosit cu permisiunea de la Am J Ophthalmol. 2001;131(5):599-606 )
epidemiologie și etiologie
|
SIGNS
|
simptome
|
tratament/management
|
- Du L, Kijlstra a, Boala Yang P. Vogt-Koyanagi-Harada: perspective noi în fiziopatologie, diagnostic și tratament. Prog Retin Ochi Res 2016; 52: 84-111. https://PubMed.gov/26875727. DOI: 10.1016/j.preteyeres.2016.02.002
- Yamaki K, Gocho K, Hayakawa K, Kondo I, Sakuragi S. proteinele familiei tirozinazei sunt antigene specifice bolii Vogt-Koyanagi-Harada. J Immunol 2000; 165 (12):7323-7329. https://PubMed.gov/11120868
- Horie Y, Takemoto Y, Miyazaki A, Namba K, Kase S, Yoshida K, Ota M, Hasumi Y, Inoko H, Mizuki N, Ohno S. Familia genei tirozinazei și boala Vogt-Koyanagi-Harada la pacienții japonezi. Mol Vis 2006; 12: 1601-1605. https://PubMed.gov/17200659
- Ng JY, Luk FO, Lai TY, Pang CP. Influența geneticii moleculare în boala Vogt-Koyanagi-Harada. J Oftalmic Inflamm Infecta 2014; 4: 20. https://PubMed.gov/25097674. DOI: 10.1186 / s12348-014-0020-1
- Bowling B. uveită. Oftalmologia clinică a lui Kanski New York, New York: Elsevier; 2016; Capitolul 11; p. 395-465.
- Yeh PT YC, Yang CH, Lin CP. Dezlipire De Retina Nonrhegmatogenous. În: Schachat AP SS, Hinton DR, Wilkinson CP, Wiedemann P,, editor. Retina lui Ryan. New York: Elsevier; 2018; capitolul 99; p. 1828-1849.
- du-te la H RK, Rao N. Vogt-boala Koyanagi-Harada. În: Schachat AP SS, Hinton DR, Wilkinson CP, Wiedemann P, editor. Retina lui Ryan. New York, New York: Elsevier; 2018; capitolul 78; p. 1505-1515.
- Riddington l, Hall AJ, Tait B, Nicholson I, sindromul Varney M. Vogt-Koyanagi-Harada la pacienții cu strămoși Vietnamezi. Aust N Z J Oftalmol 1996; 24 (2): 147-149. https://PubMed.gov/9199747
- Sugita S, Takase H, Kawaguchi T, Taguchi C, Mochizuki M. Reacția încrucișată între peptidele tirozinazei și antigenul citomegalovirusului de către celulele T de la pacienții cu boală Vogt-Koyanagi-Harada. Int Ophthalmol 2007; 27 (2-3):87-95. https://PubMed.gov/17253112. DOI: 10.1007 / s10792-006-9020-y
- Freund BK SD, Mieler WF, Yannuzzi LA. Inflamație. Atlasul Retinei. New York, New York: Elsevier 2017; Capitolul 4; p. 279-398.
- boala Rao N. Vogt-Koyanagi-Harada. În: J YMaD, editor. Oftalmologie. New York, New York: Elsevier; 2014; capitolul 7.17; p. 761-763.
- Rao NA, Xu S, Font RL. Oftalmie simpatică. Un studiu imunohistochimic al celulelor epiteliale și gigantice. Oftalmologie 1985; 92(12):1660-1662. https://PubMed.gov/4088616
- Nussenblatt RB. Sindromul Vogt-Koyanagi-Harada. În: Whitcup RBNaSM, editor. Uveita: fundamente și Practică Clinică. Ediția a 4-a ed: Elsevier; 2010; capitolul capitolul 24.
- Citește RW, Olanda GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L, Pivetti-Pezzi P, Tessler HH, Usui M. criterii de diagnostic revizuite pentru boala Vogt-Koyanagi-Harada: raportul unui comitet internațional pentru nomenclatură. Am J Oftalmol 2001; 131 (5):647-652. https://PubMed.gov/11336942
- Chung H, Choi DG. Analiza clinică a uveitei. Coreeană J Oftalmol 1989; 3(1):33-37. https://PubMed.gov/2795939. DOI: 10.3341/kjo.1989.3.1.33
- Abu El-Asrar AM, al-Kharashi AS, Aldibhi H, Al-Fraykh H, kangave D. Vogt-Koyanagi-boala Harada la copii. Ochi (Lond) 2008;22(9):1124-1131. https://PubMed.gov/17479116. DOI: 10.1038 / sj.ochi.6702859
- Martin TD, Rathinam SR, Cunningham ET. Prevalența, caracteristicile clinice și cauzele pierderii vederii la copiii cu boala Vogt-Koyanagi-Harada din sudul Indiei. Retină 2010; 30(7):1113-1121. https://PubMed.gov/20168275. DOI: 10.1097 / IAE.0B013E3181C96A87
- Forster DJ, Verde RL, Rao NA. Manifestarea unilaterală a sindromului Vogt-Koyanagi-Harada la un copil de 7 ani. Am J Oftalmol 1991; 111 (3):380-382. https://PubMed.gov/2000916
- Yamamoto y, Fukushima A, Nishino K, Koura Y, Komatsu t, Ueno H. Vogt-Koyanagi-boala harada cu debut la pacienții vârstnici cu vârsta cuprinsă între 68 și 89 de ani. Jpn J Oftalmol 2007; 51 (1):60-63. https://PubMed.gov/17295144. DOI: 10.1007 / s10384-006-0379-0
- Wang Y, Chan CC. Diferențele de gen în boala vogt-Koyanagi-harada și oftalmia simpatică. J Oftalmol 2014; 2014: 157803. https://PubMed.gov/24734166. DOI: 10.1155/2014/157803
- Nakao K, Abematsu N, Mizushima Y, Sakamoto T. umflarea discului Optic în boala Vogt-Koyanagi-Harada. Invest Oftalmol Vis Sci 2012; 53 (4): 1917-1922. https://PubMed.gov/22408010. DOI: 10.1167 / iovs.11-8984
- Rao NA, Gupta A, Dustin L, Chee SP, Okada AA, Khairallah M, Bodaghi B, Lehoang P, Accorinti M, Mochizuki M, Prabriputaloong T, Citește RW. Frecvența caracteristicilor clinice distinctive în boala Vogt-Koyanagi-Harada. Ophthalmology 2010;117(3):591-599, 599.e591. https://PubMed.gov/20036008. DOI: 10.1016/j.ophtha.2009.08.030
- Veerappan M, Fleischman D, Ulrich JN, Stinnett SS, Jaffe GJ, Allingham RR. The Relationship of Vogt-Koyanagi-Harada Syndrome to Ocular Hypertension and Glaucoma. Ocul Immunol Inflamm 2017;25(6):748-752. https://PubMed.gov/27438521. DOI: 10.1080/09273948.2016.1189578
- Baltmr A, Lightman S, Tomkins-Netzer O. Vogt-Koyanagi-Harada syndrome – current perspectives. Clin Ophthalmol 2016;10:2345-2361. https://PubMed.gov/27932857. DOI: 10.2147/OPTH.S94866
- Kitaichi n, Matoba H, Ohno S. rolul pozitiv al puncției lombare în diagnosticul bolii Vogt-Koyanagi-Harada: subseturi de limfocite în umoarea apoasă și lichidul cefalorahidian. Int Ophthalmol 2007; 27 (2-3):97-103. https://PubMed.gov/17211585. DOI: 10.1007 / s10792-006-9016-7
- Oshima Y, Harino s, Hara Y, Tano Y. constatări angiografice verzi Indocianine în boala Vogt-Koyanagi-Harada. Am J Oftalmol 1996; 122 (1):58-66. https://PubMed.gov/8659599
- Citește RW, Yu F, Accorinti M, Bodaghi B, Chee SP, Fardeau C, du-te la H, Olanda GN, Kawashima H, Kojima E, Lehoang P, Lemaitre C, Okada AA, Pivetti-Pezzi P, Secchi a, Vezi RF, Tabbara KF, Usui M, Rao NA. Evaluarea efectului asupra rezultatelor căii de administrare a corticosteroizilor în boala acută Vogt-Koyanagi-Harada. Am J Oftalmol 2006; 142 (1):119-124. https://PubMed.gov/16815259. DOI: 10.1016 / j. ajo.2006.02.049
- Rubsamen PE, Gass JD. Sindromul Vogt-Koyanagi-Harada. Curs clinic, terapie și rezultat vizual pe termen lung. Arch Ophthalmol 1991;109(5):682-687. https://PubMed.gov/2025171
- Urzua ca, Velasquez V, Sabat P, Berger O, Ramirez S, Goecke A, V Inksquez DH, Gatica H, Guerrero J. tratamentul imunomodulator anterior este asociat cu rezultate vizuale mai bune la un subset de pacienți cu boala Vogt-Koyanagi-Harada. Acta Ophthalmol 2015; 93(6): e475-480. https://PubMed.gov/25565265. DOI: 10.1111 / aos.12648
- Citește RW, Rechodouni A, Butani n, Johnston R, LaBree LD, Smith RE, Rao NA. Complicații și factori de prognostic în boala Vogt-Koyanagi-Harada. Am J Oftalmol 2001; 131 (5):599-606. https://PubMed.gov/11336934
format de citare sugerat
Mai AP, Tran C, Wilson CW, Fox ar, Boldt HC. Boala Vogt-Koyanagi-Harada (VKH). EyeRounds.org. 1 Aprilie 2019. Disponibil de la http://EyeRounds.org/cases/284-vogt-koyanagi-harada.htm
Leave a Reply