Vogt-Koyanagi-Harada (VKH) Doença
Autores: Anthony P. Mai, BS; Charlene Tran, BS; Caroline W. Wilson, MD; Austin R. Fox, MD; H. Culver Boldt, MD
1º de abril, 2019
APRESENTAÇÃO INICIAL
Queixa principal
a visão Embaçada e dores de cabeça
História do Presente Doença
A 44 anos Vietnamita mulher apresentada ao departamento de emergência com 10 dias de histórico de progressiva visão embaçada em ambos os olhos, de um a três dias de histórico de dores de cabeça severas. Sua perda de visão central não tinha melhorado com uma refração por seu optometrista. Suas graves dores de cabeça occipital pioraram com o movimento e foram associadas com mal-estar generalizado, fadiga extrema, fotofobia leve e rasgo. O acetaminofeno aliviou parcialmente a dor. ela tinha viajado recentemente para o Vietname, mas negou ter encontrado contactos doentes lá. Ela negou a claudicação da mandíbula, febres ou alterações de peso. Ela negou erupções cutâneas, alterações auditivas, zumbido, tonturas, dormência ou formigueiro. Ela negou alguma vez ter tuberculose. Não tinha antecedentes de problemas de visão, doenças auto-imunes ou cancro.
Passado Ocular da História
- História da cirurgia da pálpebra cosméticos (bilateral blefaroplastia) três anos antes de
- Sem história de trauma ocular ou doença
História Médica
Nenhum
Medicamentos
o Paracetamol como necessário
Alergias
Não conhecido alergias a medicamentos
a História da Família
Sem histórico de doença ocular ou doença auto-imune
História Social
Ela emigrou do Vietnã, vários anos antes da apresentação. Ela é casada e tem três filhos. Ela trabalha num salão de manicura. Ela não consome produtos de tabaco, álcool ou substâncias ilícitas. Viaja para o Vietname de seis em seis a doze meses.
a Revisão dos Sistemas
Negativo, exceto para o que está detalhado na história do presente doença
OCULAR EXAME
a Acuidade Visual com e sem correção (Snellen)
- olho Direito (OD): 20/300 (nenhuma melhoria com pinhole)
- olho Esquerdo (OS): 20/60-2+2 (nenhuma melhoria com pinhole)
Motilidade Ocular/Alinhamento
Cheio de extraocular movimentos em ambos os olhos (OU)
a Pressão intra-ocular (IOP): (Tonopen)
- OD: 12 mm hg
- OS: 14 mmHg
Alunos
- OD: 4 mm no escuro, 3 mm na luz, não em relação defeito pupilar aferente (RAPD)
- OS: 4 mm no escuro, 3 mm na luz, não RAPD
o Confronto visual campos: (Contagem de dedos)
- OD: Central escótomos bilaterais
- OS: Total inferotemporal defeito
Externo
Normal em ambos os lados
lâmpada de Fenda exame
- Tampas/cílios: Normal UO
- Conjuntiva/esclera: Clara e tranquila UO
- Córnea: 1+ punctate erosões epiteliais, não keratic precipita UO
- câmara Anterior: Rastreamento de celular e reflexos e profunda UO
- Iris: Normal arquitetura UO
- Lente: Claro UO
Dilatada fundo de exame (DFE)
- Vítreo: o traçado anterior vítreo células UO
- Disco:
- OD: Grau 3 disco de edema, hyperemic
- sistema operacional: Grau 2-3 disco edema, hyperemic
- Copo-para-disco razão: 0.0 UO
- Macula:
- OD: 3+ cystoid edema macular (CME) e subretinal fluido (SRF) se estendendo a partir do disco para o temporal macula. Sem lípidos ou exsudados. Coróide de aspecto Boggy.
- OS: 2+ CME e SRF estendendo-se do disco através da fovea. 1-2+ lípidos lineares que se estendem do disco para fovea. Coróide de aspecto Boggy.Navios:
- od: Embainhamento temporalmente
- Aguda posterior multifocal placoid pigmento epitheliopathy (APMPPE)
- Central serosa chorioretinopathy
- neurite Óptica
- Panuveitis
- auto-imune (por exemplo, LÚPUS, sarcoidose)
- a Infecção (e.g., a sífilis, a tuberculose, a Bartonella henselae)
- Malignidade (por exemplo, linfoma ocular)
- Posterior scleritis
- oftalmia Simpática
- efusão Uveal síndrome
- Vogt-Koyanagi-Harada Síndrome
- Prodromal: esta fase inicial pode apresentar-se como uma doença do tipo gripal com sintomas predominantemente constitucionais, tais como cefaleias, tonturas, febre, fadiga e/ou náuseas. Foram relatados sintomas neurológicos de meningite, paralisia do nervo craniano e neurite óptica, bem como sintomas auditivos de zumbido, disacusia e vertigens . Fotofobia, visão turva, flutuantes e/ou dor ocular geralmente começam dentro de 48 horas após sintomas prodromais . A fase prodromal geralmente dura de alguns dias a semanas.
- Uveítico agudo: esta fase inclui visão desfocada, fotofobia, injecção conjuntival e dor ocular. Pode haver uveíte anterior ligeira que à primeira vista não parece granulomatosa. Início Unilateral tipicamente transições para envolvimento bilateral em 1-2 semanas. Pode desenvolver-se uveíte anterior granulomatosa com precipitados queráticos de gordura de carneiro. Os resultados do exame Posterior podem incluir edema e hiperemia do nervo óptico, áreas multifocais de coroidite, múltiplas áreas de destacamentos serosos da retina localizadas no fundo posterior, espessamento coroidal, dobras corioretinais radiantes e vitrite . Destacamentos serosos da retina podem formar um padrão de cloverleaf no fundo posterior e podem progredir para destacamentos extensos em casos graves . O glaucoma inflamatório agudo tem sido associado a esta fase da doença e pode apresentar-se com uma câmara anterior superficial secundária ao edema corporal ciliar, imitando o fechamento do ângulo agudo . A duração da fase uveítica aguda depende do diagnóstico e tratamento imediato. este estágio Geralmente se desenvolve várias semanas após a fase aguda e é caracterizado por vitiligo (por exemplo, face, mãos, ombros ou costas), poliose e alopecia . Despigmentação perto do limbo da córnea, conhecido como sinal de Sugiura, pode ser visto um mês após o início da doença ; no entanto, este sinal é raramente visto fora da população japonesa . A despigmentação coroidal geralmente ocorre ao longo de alguns meses e resulta na cor laranja-vermelha brilhante do coróide e o clássico “sunset glow fundus”.”Sunset glow fundus é considerado o mais importante e preditivo no diagnóstico de VKH crônica . Cicatrizes corioretinais bem definidas, redondas e nummulares podem formar-se na periferia média. A fase uveítica crónica dura normalmente vários meses.
- crónica-recorrente: Esta fase é caracterizada por episódios recorrentes de granulomatosa uveíte anterior com carne de carneiro a gordura keratic precipitados, iris nódulos, iris despigmentação, posterior synechiae, cataracts subcapsular posterior, glaucoma secundário, choroidal neovascular membranas, e, em última análise, subretinal fibrose e o sholud chorioretinal atrofia . A fase crônica tipicamente se desenvolve pelo menos seis meses após a apresentação inicial. Os destacamentos serosos da retina presentes durante as fases aguda e convalescente normalmente não reaparecem com o tratamento agressivo com corticosteróides .
- Com equívocos fundo resultados; ambos dos seguintes devem estar presentes também:
Os: Normal
periferia:
- od: tufo da retina cística anterior ao equador às 10: 30 OS: raso SRF anterior ao equador às 4::00
 |
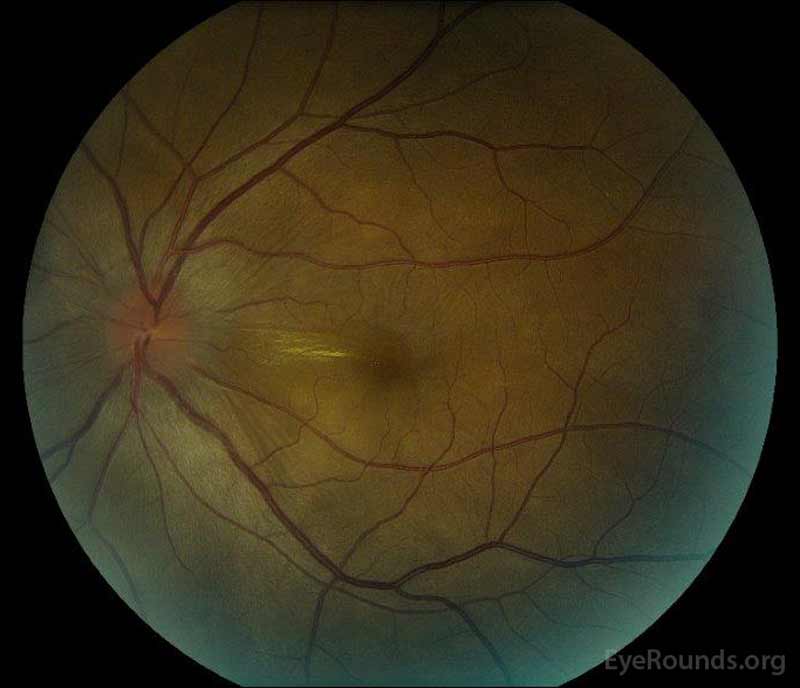 |
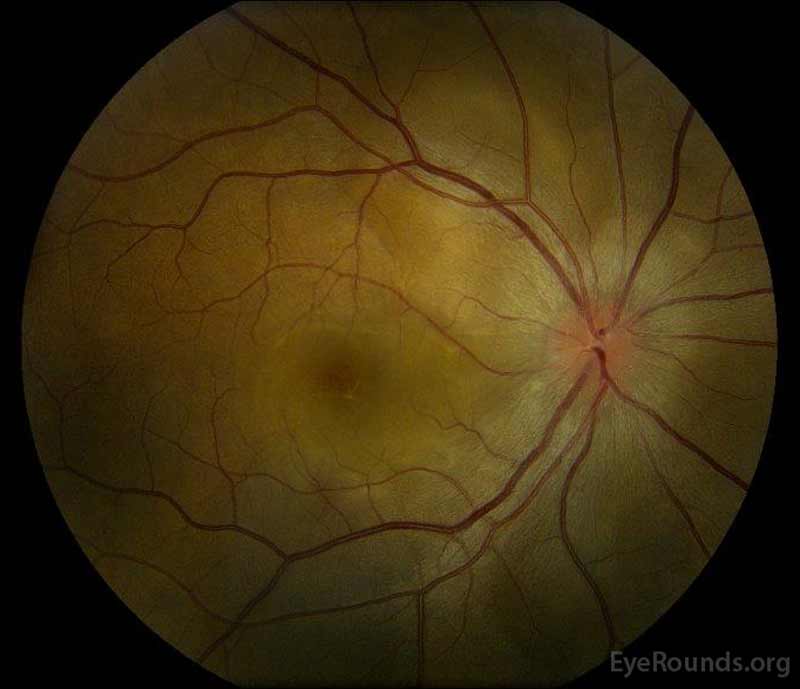
Figura 1: a Cor de fundo de fotografias a apresentação de: (imagem à Esquerda) O olho direito tem disco de edema e leve hiperemia bem como subretinal fluido estendendo-se a partir do disco temporalmente através da mácula. Há também um descolamento seroso focal retinal superotemporal para o disco, ao longo da arcada superior. (Right image) The left eye has disc edema and mild hyperemia, along with subretinal fluid extending from the disc to the macula and linear lipid deposits in the nasal macula.
 |
 |
 |
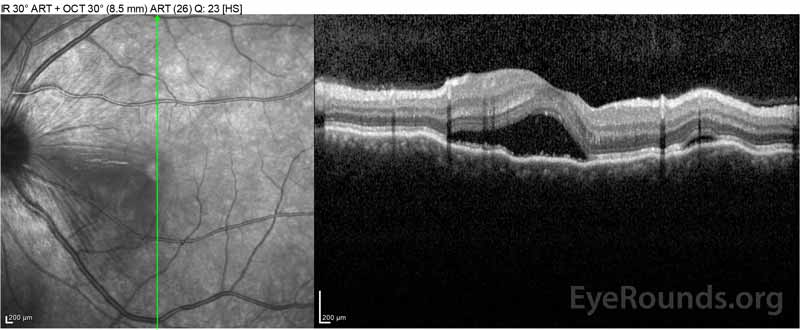 |
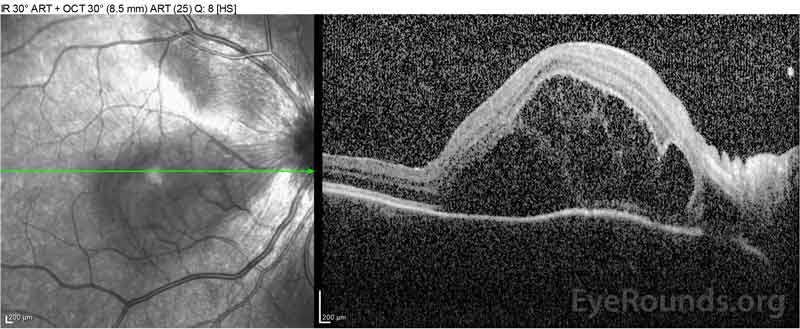
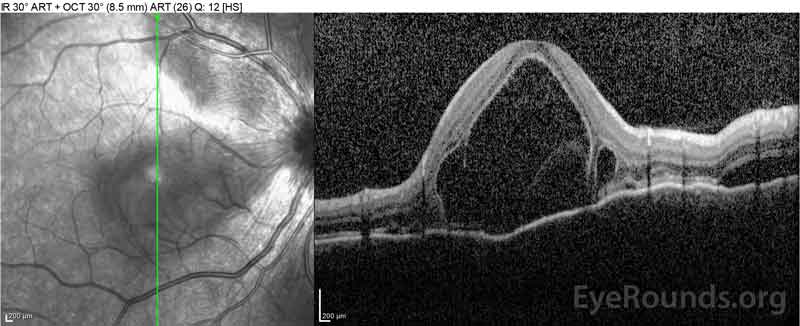
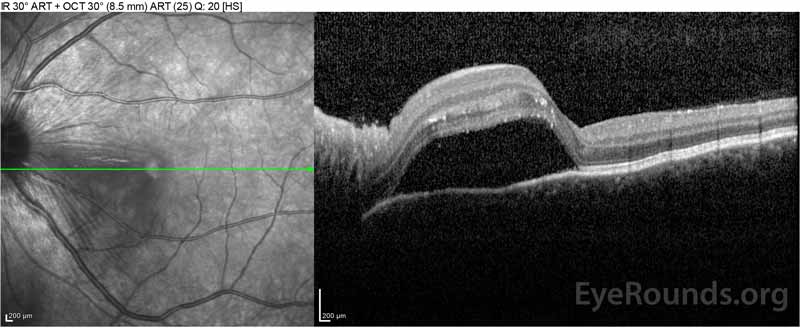
Figure 2: A tomografia de coerência óptica (OCT) do olho direito (painéis superiores) mostra um descolamento seroso da retina envolvendo a fovea com fluido intretinal extenso, ruptura das camadas externas da retina, e ondulações do coróide espessado. O OCT do olho esquerdo (painéis inferiores) mostra um descolamento seroso da retina na mácula nasal estendendo-se até à fovea.
 |
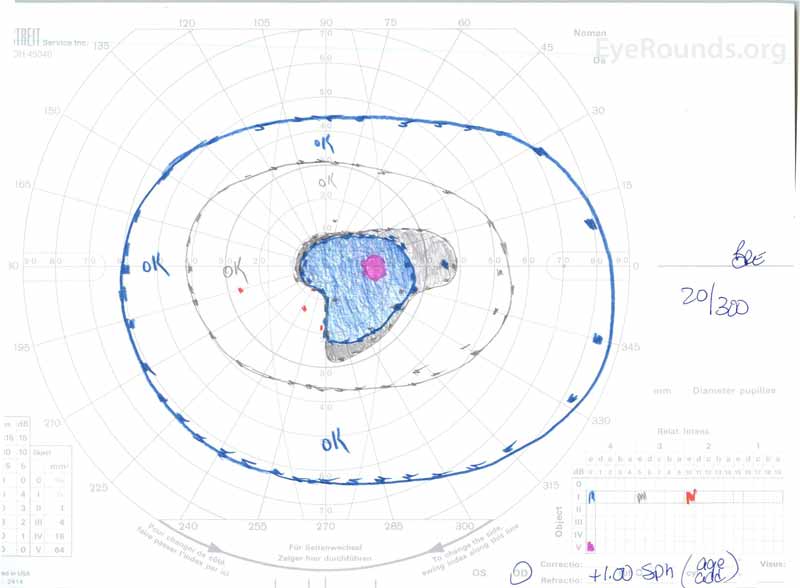 |
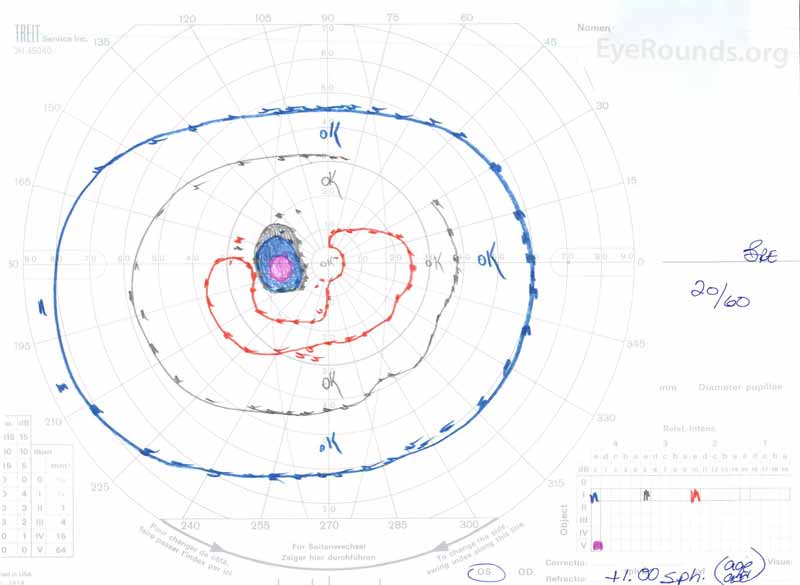
Figura 3: Goldman visual fields (GVF), OU. O sistema operacional mostra um ponto cego fisiológico aumentado e um ligeiro scotoma central. A do Mostra um scotoma central moderadamente denso, incorporando o ponto cego fisiológico e estendendo-se inferotemporalmente.
B-scan: Sem sinais de scleritis, leve vitreal opacidades/células inferiormente
o Diagnóstico Diferencial
WORK-UP
hemograma Completo
contagem de glóbulos Brancos: 4.9 K/mm3 (Ref: 3.7-10.5)
a contagem de células Vermelhas 3.99 M/mm3 (Ref: 4.0-5.2)
a Hemoglobina a 11,6 g/dL (Ref: 11.9-15.5)
Hematócrito 35 % (Ref.: 35-47)
Basic metabolic panel
Sodium 138 mEq/L (Ref: 135-145)
Potassium 4.3 mEq/L (Ref: 3.5-5.0)
Chloride 107 mEq/L (Ref: 95-107)
CO2 20 mEq/L (Ref: 22-29)
Blood urea nitrogen 16 mEq/dL (Ref: 10-20)
Creatinine 0.7 mg/dL (Ref: 0.5-1.0)
C-reactive Protein (CRP): <0.5 mg/dL (Ref: <=0.5)
Erythrocyte sedimentation rate (ESR): 12 mm/Hr (Ref: 0-20)
Angiotensin–converting enzyme (ACE): 13 U/L (Ref: 8-52)
QuantiFERON ® -TB Gold: negativo
Ferro, sangue 54 microgramas/dL (Ref: 37-145)
Total de ligação do ferro capacidade 379 microgramas/dL (Ref: 250-425)
CURSO CLÍNICO
O paciente foi inicialmente avaliada pelo departamento de emergência dada a sua denúncias do surgimento de fortes dores de cabeça e perda de visão. Tomografia computadorizada cerebral (TC) e ressonância magnética (MRI) exames não foram comuns. Os ESR e os CRP estavam dentro dos níveis normais. A clínica de Oftalmologia avaliou – a no dia seguinte e encontrou destacamentos serosos bilaterais da retina e panuveíte. ACE e Quantificon-TB Gold labs foram ambos negativos. Ela foi diagnosticada com a doença de Vogt-Koyanagi-Harada com base em sua apresentação clínica e descendência asiática. Ela foi tratada com 80 mg de prednisona por dia, acetaminofeno conforme necessário para dores de cabeça, e suplementos de vitamina D e cálcio. Suas dores de cabeça rapidamente se resolveram, e sua acuidade visual melhorou constantemente ao longo das duas semanas seguintes. A dose de prednisona foi então reduzida para 40 mg ao longo de três semanas, com a resolução contínua dos sintomas e melhoria da acuidade visual. Não teve recorrência de dores de cabeça ou agravamento da visão durante a lapidação da prednisona. Na sua consulta mais recente, ela tinha baixado para 5 mg em dias alternados, sem retorno dos sintomas. Sua acuidade visual na visita de acompanhamento foi de 20/15-2 DOD e 20/20+2 OS, e o OCT macular mostrou resolução completa de edema do disco e destacamentos serosos da retina em ambos os olhos (Figura 4).
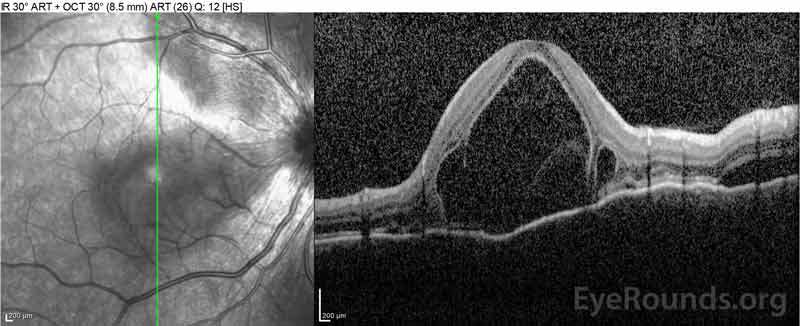
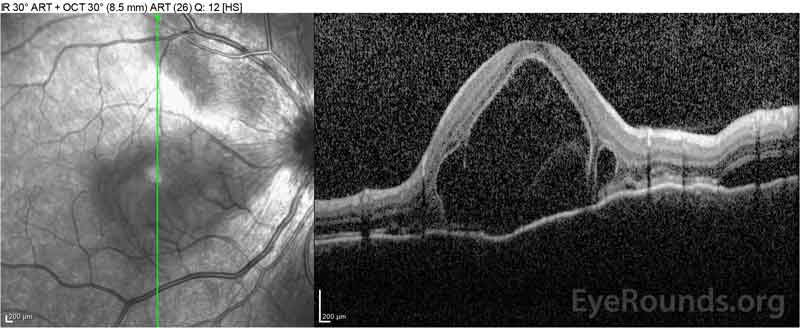
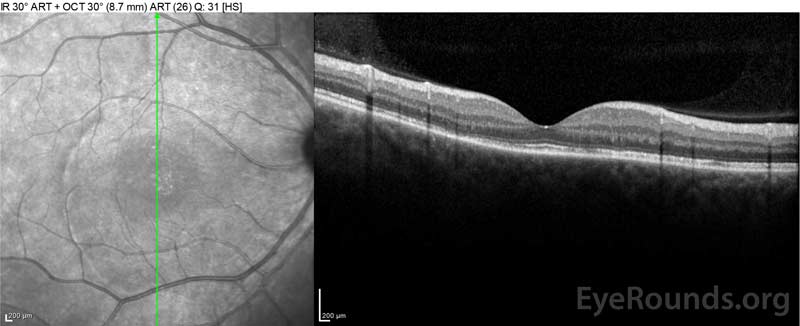
Figura 4: Tomografia de coerência óptica que mostra o fluido subretinal na linha de base (topo) e o curso de resolução à uma semana (meio) e cinco semanas (fundo) enquanto em uma dose oral de prednisona de alta dose. Repare na suavização das ondulações coroidais com o tratamento.
 |
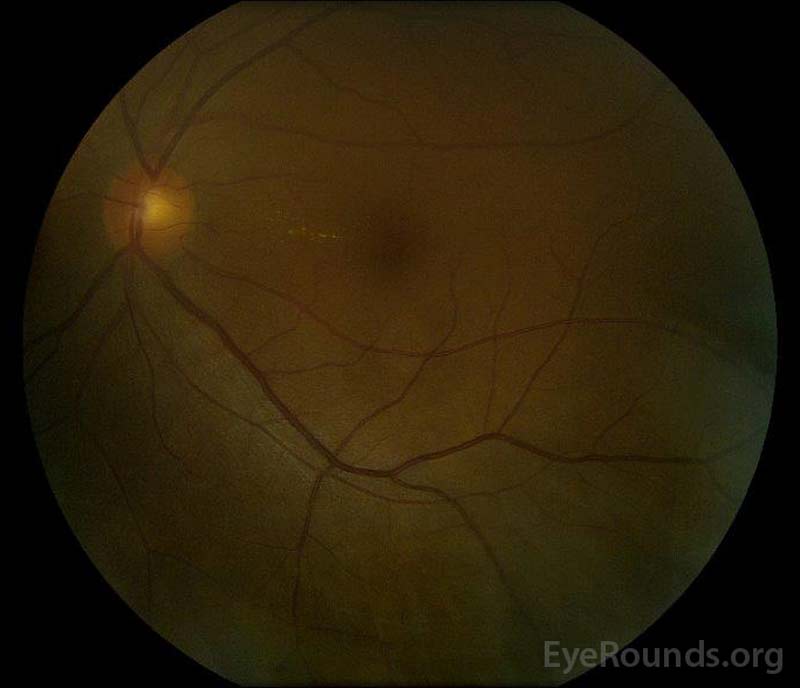 |
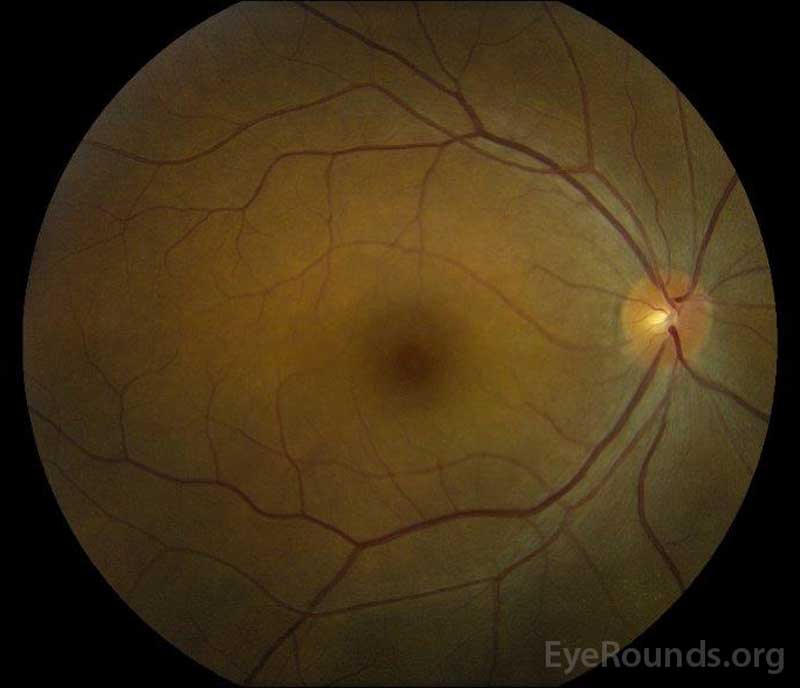
Figura 5: Fotografia de fundo de cor dos olhos direito (a) e esquerdo (B) durante a fase de convalescença demonstrando melhoria no fluido subretinal e edema de disco.
o DIAGNÓSTICO
Incompleta Vogt-Koyanagi-Harada Doença
DISCUSSÃO
Vogt-Koyanagi-Harada (VKH) a Doença é uma condição auto-imune sistêmica caracterizada por bilateral não-granulomatosa necrotizante panuveitis associados com extraocular tegumentar alterações, tais como poliosis e vitiligo, e a inflamação que afetam a úvea, ouvido interno, do cabelo e das meninges. A doença de Harada é a uveíte isolada sem os sinais sistêmicos ou sintomas de VKH.
etiologia
a etiologia da doença de VKH ainda é largamente desconhecida, apesar dos esforços de investigação em curso. Pensa-se que seja uma doença auto-imune adquirida envolvendo hipersensibilidade mediada pelas células T aos Auto-antigénios melanocíticos, com uma predisposição genética subjacente e possível gatilho microbiano . A tirosinase e os peptídeos relacionados com a tirosinase são antigénios melanócitos que têm sido sugeridos como alvos de processos auto-imunes na VKH . No entanto, o aumento do risco de doença VKH não foi associado à família genética da tirosinase, de acordo com um estudo .devido ao aumento da prevalência entre certos grupos étnicos e de gênero, acredita-se que haja uma predisposição genética na patogênese da VKH. Múltiplos genes, incluindo os genes do antigénio leucocitário humano (HLA) e da interleucina (IL), têm sido associados à VKH em diferentes populações étnicas . Os receptores HLA são complexos histocompatíveis principais em humanos que apresentam peptídeos ao sistema imunológico. HLA-DR1, HLA-DR4, HLA-DRB1*0405, e HLA-DRw53 são vários haplotipos encontrados em pacientes com doença VKH . HLA-DR4 é mais comum em japoneses e hispânicos,enquanto HLA-DRB1 * 0405 é mais frequente em pacientes coreanos e do oriente médio . Ambos os alelos HLA-DR4 e HLA-DRB1*0405 são encontrados em pacientes Vietnamitas . O receptor HLA-DRB1 liga-se aos antigénios melanócitos em capacidades variáveis. Apesar destas associações, os ensaios genéticos não são recomendados neste momento.
Dada a habitual sintomas prodrômicos que acompanham VKH, incluindo febre, dores de cabeça, meningismus e zumbido, um incitação de etiologia viral tem sido sugerida como um gatilho para VKH início, através de mecanismos de mimetismo molecular em pacientes geneticamente predispostos. A glicoproteína h do envelope do citomegalovírus tem uma homologia significativa de aminoácidos ao péptido da tirosinase, e a infecção por CMV pode desencadear a VKH através do mimetismo molecular (ou seja, o reconhecimento pelos receptores da classe II do HLA) . Ebstein-bar virus (EBV) também foi implicado. No entanto, não há evidências definitivas sobre a etiologia viral da VKH, e permanece incerto o que desencadeia a resposta autoimune da VKH .
patofisiologia
há quatro fases clássicas do VKH que podem ter apresentações variáveis: pródromal, uveítico agudo, convalescente e crónico-recorrente. As alterações histopatológicas normalmente começam na fase aguda . a fase uveítica aguda é caracterizada por espessamento uveal bilateral secundário à inflamação granulomatosa. Os granulomas consistem em linfócitos, macrófagos e células epitelióides e gigantes cheias de grânulos . Apesar das células epitelióides terem sido previamente consideradas melanócitos alterados, um estudo imunohistoquímico de seguimento sugeriu uma origem dos macrófagos tecidulares . Granulomas cheios de histiócitos epitelioides, chamados nódulos de Dalen-Fuchs, podem muitas vezes ser vistos entre o pigmento retinal epitélio (RPE) e a membrana de Bruch. A inflamação granulomatosa uveal conduz a espessamento coroidal e a destacamentos exsudativos da retina cheios de fluido proteináceo. Além disso, a pleocitose(I.E., aumento da contagem de células) pode estar presente na câmara anterior e vítreo .a fase convalescente é identificada pela despigmentação das áreas coróide e extraocular, incluindo a pele periocular e o cabelo. Um coróide depigmentado colocado contra um nervo óptico pálido dá a impressão de um fundo” sunset-glow”, que é uma característica clássica desta fase de VKH . Além disso, os nódulos de Dalen-Fuchs tornam-se mais proeminentes sob o RPE na fase convalescente .
a fase crónica recorrente caracteriza-se pela diminuição da espessura coroidal, resolução dos destacamentos serosos da retina, vitrite crónica ligeira e inflamação recorrente do segmento anterior granulomatoso. A Neovascularização coroidal (CNV) e a fibrose subretinal podem desenvolver-se durante esta fase e são indicadores de progressão grave da doença . Cataratas e glaucoma secundário são outras complicações da inflamação de longa data ou recorrente nesta fase .
Epidemiologia
VKH é prevalente em raças com pigmento de pele mais escura, especialmente asiáticos, sul-Americanos, Médio Oriente e Nativos Americanos. A doença de VKH é responsável por>10% da uveíte nestas populações . Apenas 1-4% dos casos de uveitis são considerados secundários à doença de VKH nos Estados Unidos (7). Nos Estados Unidos, a maioria dos casos de VKH foram encontrados para afetar indivíduos asiáticos, hispânicos e/ou nativos americanos decentes . Curiosamente, a doença de VKH raramente afeta africanos apesar de sua pigmentação escura . A incidência da doença de VKH varia muito entre os subgrupos raciais nos países vizinhos . Por exemplo, a incidência de VKH na Coreia é de apenas 2%, muito menor do que a encontrada no Japão e na China .
VKH tem um início típico de 20 a 50 anos de idade ; no entanto, estudos sugerem que 3,1-13,4% dos casos de VKH são pacientes pediátricos e 10% dos casos são ≥65 anos de idade . Classicamente, pensa-se que a VKH tem uma predileção pelo sexo feminino, e enquanto a maioria dos estudos mostram que a VKH afeta desproporcionalmente as mulheres, alguns estudos têm mostrado uma predisposição masculina ou nenhuma predisposição de gênero .como acima mencionado, os quatro estágios da doença de VKH são pródromal, uveítico, convalescente e recorrente crônica. Cada etapa apresenta características clínicas distintas.
critérios de diagnóstico
os critérios de diagnóstico mais recentes, chamados de critérios de diagnóstico revistos (RDC) para VKH, foram definidos em 1999 na primeira oficina internacional em VKH . Estes são descritos no quadro 1. A RDC é útil na medida em que divide a VKH em três diferentes categorias diagnósticas baseadas na fase da doença durante a qual um paciente apresenta: completo, incompleto e provável. Esta categorização da doença permite uma gestão adequada e precoce da doença ” provável “que pode ajudar a prevenir a progressão para a doença” completa”. são essenciais trabalhos para outras causas de inflamação ocular, tanto infecciosa como auto-inflamatória. Estes podem incluir a taxa de sedimentação eritrocitária (ESR), proteína C-reactiva (CRP), teste de quantificon-Gold para a tuberculose, reagina plasmática rápida (RPR) para a sífilis, enzima de conversão da angiotensina (ECA) e raio-X torácico para a sarcoidose, anticorpo antinuclear (ANA) e P-/C-ANCA. Além disso, um histórico de trauma ocular recente ou cirurgia intra-ocular deve ser notado e provavelmente sugere oftalmia simpática (SO) como o diagnóstico mais provável dada a apresentação muito semelhante e patofisiologia compartilhada entre SO e VKH .
para suportar um diagnóstico de VKH em casos equívocos, uma punção lombar pode ser realizada para procurar por pleocitose linfocítica e monocítica; no entanto, isso raramente é empregado clinicamente. Oitenta por cento dos pacientes têm pleocitose no líquido cefalorraquidiano (LCR) em uma semana e 97% têm pleocitose em três semanas. O aumento dos níveis de células imunitárias pode durar até oito semanas após o início da doença . Os perfis de marcadores da superfície das células T são semelhantes entre o líquido cefalorraquidiano e o humor aquoso, mas diferentes do sangue. Isto sugere a capacidade do LCR para refletir com precisão a inflamação uveal na doença de VKH .
Tabela 1. Critérios de diagnóstico revistos para a doença de Vogt-Koyanagi-Harada
*Da Tabela 1 em (15). doença de Vogt-Koyanagi-Harada completa (devem estar presentes os critérios 1 a 5) nenhuma evidência clínica ou laboratorial sugestiva de outras entidades da doença ocular. envolvimento ocular Bilateral (a ou b devem ser cumpridos, dependendo da fase da doença quando o doente é examinado). manifestações precoces da doença. deve haver evidência de coroidite difusa (com ou sem uveíte anterior, reacção inflamatória vítrea ou hiperemia do disco óptico), que pode manifestar-se como uma das seguintes:
- áreas Focais de atraso no choroidal perfusão, áreas multifocais de pinpoint de fuga, grande placoid áreas de hyperfluorescence, o agrupamento dentro de subretinal fluido, nervo óptico e coloração (listadas em ordem seqüencial aparência) por angiografia com fluoresceína e
- Difusa choroidal espessamento, sem evidência de posterior scleritis por ultra-sonografia. manifestações tardias da doença. história sugestiva de presença prévia de achados de 3a, e tanto (2) e (3) abaixo ou sinais múltiplos de (3): despigmentação Ocular (uma das seguintes manifestações é suficiente): (a) fundo de brilho do pôr-do-sol, ou (B) sinal de Sugiura. outros sinais oculares: aglomeração e/ou migração do epitélio pigmento da retina, ou uveíte anterior recorrente ou crónica. os resultados neurológicos/auditivos podem ter desaparecido durante o exame. meningismo (mal-estar, febre, dores de cabeça, náuseas, dor abdominal, rigidez do pescoço e costas, ou uma combinação destes factores; contudo, a dor de cabeça por si só não é suficiente para satisfazer a definição de meningismo), ou
zumbido, ou pleocitose do líquido cefalorraquidiano. achado Integumentário (Não anterior ao início do sistema nervoso central ou doença ocular). Alopécia, ou poliose, ou Vitiligo. doença de Vogt-Koyanagi-Harada incompleta (devem estar presentes os critérios 1 a 3 e 4 ou 5)resultados neurológicos/auditivos; conforme definido para a doença completa de Vogt-Koyanagi-Harada acima ou para os achados Integmentários; como definido para a doença completa de Vogt-Koyanagi-Harada acima.doença provável de Vogt-Koyanagi-Harada (doença ocular isolada; devem estar presentes critérios 1 a 3) não há história de trauma ocular penetrante ou cirurgia anterior ao início inicial da uveíte.nenhuma evidência clínica ou laboratorial sugestiva de outras entidades da doença ocular.envolvimento ocular Bilateral, como definido acima para a doença de Vogt-Koyanagi-Harada. ”
Testing/Laboratory-Up
no trabalho inicial de VKH, deve-se considerar a obtenção dos seguintes testes:
- tomografia de coerência óptica (OCT): Na fase uveítica aguda, a OCT irá provavelmente apresentar espessamentos coroidais significativos e destacamentos serosos da retina. A acumulação de fluido subretinal pode ter septações que se acredita serem membranas fibrina e produtos inflamatórios, criando uma estrutura lobular que também pode ser visto na angiografia fluoresceína. Na fase convalescente, o OCT pode detectar áreas de afinação da retina após inflamação resolvida após tratamento com corticosteróides .ultra-sonografia de varrimento B: Na fase aguda, a ultrassonografia pode apresentar espessamento coroidal posterior difuso, espessamento escleral posterior, destacamentos retinianos e opacidades vítreas . Podem observar-se efusões ciliares com ecografia biomicroscópica . Este teste também é útil para descartar esclerite posterior. angiografia por fluoresceína (FA): classicamente, a FA revela pontos hipofluorescentes coroidais multifocais na fase inicial, seguidos de várias áreas hiperfluorescentes focais com fuga difusa na fase final . O corante vaza através do RPE e se acumula no espaço subretinal em torno dos pontos hiperfluorescentes. FA pode ser útil em diagnóstico quando a doença de VKH se apresenta sem sintomas extra-oculares. A hiperfluorescência do disco óptico e os defeitos da janela causados por cicatrizes corioretinais atróficas podem ser observados na periferia média . A AF na fase crónica-recorrente da doença de VKH mostra defeitos da janela não específicos devido a danos no RPE, neovascularização coroidal e fibrose subretinal .angiografia verde Indocianina (ICG) : A fase inicial do ICG mostra vasos estromais hiperfluorescentes que indicam vasculopatia coroidal e pontos escuros hipofluorescentes que correspondem a granulomas e enchimento retardado da vasculatura coroidal . A fase final revela padrões vasculares estromais difusos e hiperfluorescência coroidal difusa. A hiperfluorescência do disco é sugestiva de doença grave. A ICGA pode detectar inflamação coroidal subclínica em estadios muito precoces ou mesmo após terapêutica sistémica .punção lombar: a pleocitose no líquido cefalorraquidiano está presente na maioria dos doentes com VKH. Punção lombar deve ser realizada no início do curso da doença, desde pleocitose pode resolver
Tratamento/Gestão/Orientações
objetivos do Tratamento em VKH incluem o diagnóstico precoce e a supressão da inflamação ativa, juntamente com a prevenção de inflamação recorrente e a visão de risco de complicações, tais como o glaucoma, bolhosa, descolamento de retina, e choroidal neovascularização. o tratamento com corticosteróides sistémicos é a terapêutica preferencial para a doença de VKH, especialmente durante a fase uveítica aguda. Foi demonstrado que a via de administração de corticosteróides (oral versus intravenosa) não afecta a acuidade visual ou a ocorrência de complicações visualmente significativas no tratamento da VKH aguda . Para a doença grave, o protocolo sugerido é administração intravenosa de metilprednisolona durante três dias, seguido de tratamento oral com dose elevada de prednisona. Na doença ligeira-moderada, a prednisona oral de dose elevada pode ser suficiente para 1-2 mg / kg / dia. A dose de esteróides deve ser gradualmente reduzida ao longo de aproximadamente seis meses para prevenir a recorrência . Tratamento precoce agressivo, juntamente com testes de AF série mostrando o desaparecimento de fuga de corante através do RPE, pode ajudar a prevenir mais progressão da doença, recorrência e manifestações extraoculares . Esteróides tópicos e cicloplégicos podem diminuir as células na câmara anterior e humor vítreo. foram utilizadas injecções intravítreas e sub-Tenónicas de triamcinolona para o controlo a curto prazo da inflamação intra-ocular durante as fases aguda ou recorrente.; estas terapêuticas locais devem ser consideradas no caso de doença recalcitrante e em doentes que toleram mal os efeitos secundários sistémicos desfavoráveis dos esteróides, dada a deterioração prolongada dos esteróides. As injecções intravítreas anti-VEGF são por vezes utilizadas para o controlo da neovascularização coroidal e em casos de descolamento foveal persistente da retina serosa . os agentes poupadores de esteróides, incluindo antimetabolitos, inibidores da calcineurina, biólogos, inibidores de TNF-alfa ou agentes citotóxicos, podem ser utilizados no tratamento da VKH e devem ser cuidadosamente monitorizados, frequentemente em coordenação com um serviço de Reumatologia . Tem havido discussões em curso sobre a utilização de agentes imunossupressores não esteróides como terapêutica de primeira linha para a doença de VKH. No entanto, um estudo recente não revelou diferenças nos resultados entre o tratamento imunomodulatório de primeira linha (TMI) precoce e o tratamento com prednisona em monoterapia . Além disso, as terapêuticas imunossupressoras e biológicas são dispendiosas e requerem uma cuidadosa avaliação pré-tratamento, bem como acompanhamento frequente das análises ao sangue para avaliar os efeitos secundários graves.
na fase crónica recorrente, a recorrência frequente pode sugerir resistência à terapêutica com corticosteróides e sugere necessidade de tratamento imunomodulatório poupador de esteróides . O agente preferido para a recorrência ou intolerância aos esteróides resistentes aos esteróides é a ciclosporina . O Infliximab, o rituximab, o adalimumab e o interferão alfa-2a são agentes biológicos que também foram utilizados no tratamento da uveíte refractária da doença de VKH.
Para tratar a uveíte anterior, muitas vezes, associado com aguda VKH, esteróides tópicos (por exemplo, acetato de prednisolona 1%) e tópica cyloplegia (por exemplo, cyclopentolate 1% ou atropina 1%) devem ser prescritos, dependendo do grau de inflamação da câmara anterior. as complicações oculares estão frequentemente associadas à doença de VKH. Dadas as múltiplas fases e variedade de apresentações em que um paciente pode apresentar com VKH, o tratamento pode ser adiado em muitos casos. Em formas graves de VKH e em recorrências, a inflamação intra-ocular pode ser difícil de controlar e pode resultar em danos estruturais. Mais de 50% dos doentes desenvolvem complicações relacionadas, incluindo cataratas, glaucoma secundário, membranas neovasculares coroidais, fibrose subretinal, ou uma combinação destas (Figura 6) .
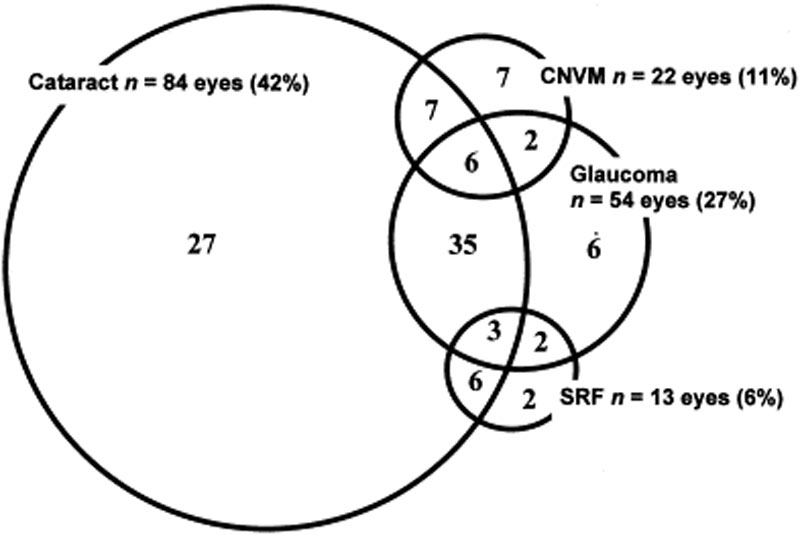
Figura 6: diagrama de Venn que mostra complicações em doentes com VKH. (Usada com permissão de Am J Ophthalmol. 2001;131(5):599-606 )
EPIDEMIOLOGIA E ETIOLOGIA
|
SIGNS
|
SINTOMAS
|
TRATAMENTO/GESTÃO
|
- Du L, Kijlstra a, Yang P. Vogt-Koyanagi-Harada disease: Novel insights into pathophysiology, diagnostic and treatment. Prog Retin Eye Res 2016; 52: 84-111. https://PubMed.gov/26875727. Doi: 10.1016/j. preteyeres.2016.02.002
- Horie Y, Takemoto Y, Miyazaki A, Namba K, Kase S, Yoshida K, Ota M, Hasumi Y, Inoko H, Mizuki N, Ohno S. Família genética da tirosinase e doença de Vogt-Koyanagi-Harada em doentes japoneses. Mol Vis 2006; 12: 1601-1605. https://PubMed.gov/17200659
- Ng jy, Luk FO, Lai TY, Pang CP. Influência da Genética molecular na doença de Vogt-Koyanagi-Harada. J Ofthalmic Inflamm Infect 2014; 4: 20. . DOI: 10.1186 / s12348-014-0020-1 Bowling B. Uveitis. Kanski’s Clinical Ophthalmology New York, New York: Elsevier; 2016; chapter 11; p. 395-465. Yeh PT YC, Yang CH, Lin CP. Descolamento Da Retina Não-Glegmatógeno. Em: Schachat AP SS, Hinton DR, Wilkinson CP, Wiedemann P, editor. A Retina do Ryan. New York: Elsevier; 2018; chapter 99; P. 1828-1849. doença de Rao N. Vogt-Koyanagi-Harada. In: Schachat AP SS, Hinton DR, Wilkinson CP, Wiedemann p, editor. A Retina do Ryan. New York, New York: Elsevier; 2018; chapter 78; P. 1505-1515. Ridgington L, Hall AJ, Tait B, Nicholson I, Varney M. Vogt-Koyanagi-Harada syndrome in patients of Vietnamese ancestry. Aust N Z J Ophthalmol 1996;24 (2): 147-149. https://PubMed.gov/9199747
- Sugita S, Takase H, Kawaguchi T, Taguchi C, Mochizuki M. Reacção cruzada entre os peptídeos da tirosinase e o antigénio do citomegalovírus pelas células T de doentes com doença de Vogt-Koyanagi-Harada. Int Ophthalmol 2007; 27 (2-3):87-95. https://PubMed.gov/17253112. DOI: 10.1007 / s10792-006-9020-y Freund BK SD, Mieler WF, Yannuzzi LA. Inflamacao. O Atlas Da Retina. New York, New York: Elsevier 2017; chapter 4; p. 279-398. doença de Rao N. Vogt-Koyanagi-Harada. In: J YMaD, editor. Oftalmologia. New York, New York: Elsevier; 2014; chapter 7.17; P. 761-763. Rao na, Xu S, Font RL. Oftalmia simpática. Um estudo imunohistoquímico de células epitelióides e gigantes. Ophthalmology 1985; 92 (12):1660-1662. https://PubMed.gov/4088616
- Nussenblatt RB. Síndrome De Vogt-Koyanagi-Harada. In: Whitcup RBNaSM, editor. Uveíte: fundamentos e Prática Clínica. 4th Edition ed: Elsevier; 2010; chapter 24.Read RW, Holland GN, Rao na, Tabbara KF, Ohno S, Arellanes-Garcia l, Pivetti-Pezzi P, Tessler HH, Usui M. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature. Am J Ophthalmol 2001; 131 (5):647-652. https://PubMed.gov/11336942
- Chung H, Choi DG. Análise clínica da uveíte. Korean J Ophthalmol 1989; 3(1):33-37. https://PubMed.gov/2795939. DOI: 10.3341 / kjo.1989.3.1.33
- Martin TD, Rathinam SR, Cunningham ET. Prevalência, características clínicas e causas de perda de visão em crianças com doença de Vogt-Koyanagi-Harada no sul da Índia. Retina 2010; 30 (7):1113-1121. https://PubMed.gov/20168275. DOI: 10.1097 / IAE.0b013e3181c96a87 Forster DJ, Green RL, Rao NA. Manifestação Unilateral da síndrome de Vogt-Koyanagi-Harada numa criança de 7 anos. Am J Ophthalmol 1991; 111 (3):380-382. https://PubMed.gov/2000916
- Yamamoto Y, Fukushima a, Nishino K, Koura Y, Komatsu T, Ueno H. Vogt-koyanagi-harada disease with onset in elderly patients aged 68 to 89 years. Jpn J Ophthalmol 2007; 51(1):60-63. https://PubMed.gov/17295144. DOI: 10.1007 / s10384-006-0379-0 Wang Y, Chan CC. Diferenças de género na doença de vogt-koyanagi-harada e oftalmia simpática. J Ophthalmol 2014; 2014: 157803. https://PubMed.gov/24734166. – Sim. 10.1155/2014/157803 Nakao K, Abematsu N, Mizushima Y, Sakamoto T. Optic disc inchaço na doença de Vogt-Koyanagi-Harada. Invest Ophthalmol Vis Sci 2012; 53 (4):1917-1922. https://PubMed.gov/22408010. DOI: 10.1167 / iovs.11-8984
- Rao NA, Gupta Um, Dustin L, Chee SP, Okada AA, Khairallah M, Bodaghi B, Lehoang P, Accorinti M, Mochizuki M, Prabriputaloong T, Leia RW. Frequência das características clínicas distintivas da doença de Vogt-Koyanagi-Harada. Ophthalmology 2010;117(3):591-599, 599.e591. https://PubMed.gov/20036008. DOI: 10.1016/j.ophtha.2009.08.030
- Veerappan M, Fleischman D, Ulrich JN, Stinnett SS, Jaffe GJ, Allingham RR. The Relationship of Vogt-Koyanagi-Harada Syndrome to Ocular Hypertension and Glaucoma. Ocul Immunol Inflamm 2017;25(6):748-752. https://PubMed.gov/27438521. DOI: 10.1080/09273948.2016.1189578
- Baltmr A, Lightman S, Tomkins-Netzer O. Vogt-Koyanagi-Harada syndrome – current perspectives. Clin Ophthalmol 2016;10:2345-2361. https://PubMed.gov/27932857. DOI: 10.2147/OPTH.S94866
- Kitaichi N, Matoba H, Ohno S. O papel positivo da punção lombar no diagnóstico de Vogt-Koyanagi-Harada doença: subconjuntos de linfócitos no humor aquoso e líquido cefalorraquidiano. Int Ophthalmol 2007; 27 (2-3):97-103. https://PubMed.gov/17211585. DOI: 10.1007 / s10792-006-9016-7 Oshima Y, Harino S, Hara Y, Tano Y. Indocianina resultados angiográficos na doença de Vogt-Koyanagi-Harada. Am J Ophthalmol 1996; 122(1):58-66. https://PubMed.gov/8659599
- Leia RW, Yu F, Accorinti M, Bodaghi B, Chee SP, Fardeau C, Goto H, Holanda GN, Kawashima H, Kojima E, Lehoang P, Lemaitre C, Okada AA, Pivetti-Pezzi P, Secchi Um, Consulte RF, Tabbara KF, Usui M, Rao NA. Avaliação do efeito nos resultados da via de administração de corticosteróides na doença aguda Vogt-Koyanagi-Harada. Am J Ophthalmol 2006; 142 (1):119-124. https://PubMed.gov/16815259. DOI: 10.1016 / j. ajo.2006.02.049
- Rubsamen PE, Gass JD. Síndrome de Vogt-Koyanagi-Harada. Curso clínico, terapia e resultado visual a longo prazo. Arch Ophthalmol 1991;109(5):682-687. https://PubMed.gov/2025171
- Urzua CA, Velasquez V, Sabat P, Berger S, Ramirez S, Goecke Um, Vásquez DH, Gatica H, Guerrero, J. Anterior imunomoduladores tratamento está associado com melhor visual resultados em um subgrupo de pacientes com Vogt-Koyanagi-Harada doença. Acta Ophthalmol 2015; 93(6):E475-480. https://PubMed.gov/25565265. DOI: 10.1111 / aos.12648
- Read RW, Rechodouni a, Butani N, Johnston R, LaBree LD, Smith RE, Rao na. Complicações e factores de prognóstico na doença de Vogt-Koyanagi-Harada. Am J Ophthalmol 2001; 131 (5):599-606. https://PubMed.gov/11336934
Yamaki K, Gocho K, Hayakawa K, Kondo i, Sakuragi S. tirosinase as proteínas da família são antigénios específicos da doença de Vogt-Koyanagi-Harada. J Immunol 2000; 165 (12):7323-7329. https://PubMed.gov/11120868
Abu El-Asrar AM, Al-Kharashi AS, Aldibhi H, Al-Fraykh H, Kangave D. Vogt-Koyanagi-Harada disease in children. Olho (Lond) 2008;22(9):1124-1131. https://PubMed.gov/17479116. DOI: 10.1038 / sj.olho.6702859
formato de citação sugerido
Mai AP, Tran C, Wilson CW, Fox AR, Boldt HC. Doença de Vogt-Koyanagi-Harada (VKH). EyeRounds.org. 1 De Abril De 2019. Disponível em http://EyeRounds.org/cases/284-vogt-koyanagi-harada.htm
Leave a Reply