Choroba Vogt-Koyanagi-Harada (VKH)
autorzy: Anthony P. Mai, BS; Charlene Tran, BS; Caroline W. Wilson, MD; Austin R. Fox, MD; H. Culver Boldt, MD
Kwiecień 1, 2019
wstępna prezentacja
Główna Skarga
niewyraźne widzenie i bóle głowy
Historia obecnej choroby
44-letnia Wietnamka zaprezentowała się na oddziale ratunkowym z 10-dniową historią postępującego niewyraźnego widzenia w obu oczach i trzydniową historią silnych bólów głowy. Jej utrata widzenia centralnego nie uległa poprawie po refrakcji przez optometrystę. Jej silne bóle potyliczne pogarszały się wraz z ruchem i były związane z uogólnionym złym samopoczuciem, skrajnym zmęczeniem, łagodną światłowstręt i łzawienie. Paracetamol częściowo złagodził ból.
niedawno podróżowała do Wietnamu, ale zaprzeczyła spotykaniu się tam z chorymi kontaktami. Zaprzeczyła chromaniu szczęki, gorączce lub zmianom masy ciała. Zaprzeczyła wysypki skórne, zmiany słuchu, szumy uszne, zawroty głowy, drętwienie lub mrowienie. Zaprzeczyła, że kiedykolwiek chorowała na gruźlicę. Nie miała wcześniej problemów ze wzrokiem, chorób autoimmunologicznych ani raka.
Historia oczna w przeszłości
- historia chirurgii plastycznej powiek (obustronna plastyka powiek) trzy lata wcześniej
- brak historii urazu lub choroby ocznej
historia choroby w przeszłości
brak
leki
Paracetamol w razie potrzeby
alergie
Brak znanych alergii na leki
historia rodzinna
brak historii choroby oczu lub choroby autoimmunologicznej
historia społeczna
wyemigrowała z Wietnamu kilka lat przed prezentacją. Jest mężatką i ma troje dzieci. Pracuje w salonie paznokci. Nie spożywa wyrobów tytoniowych, alkoholu ani nielegalnych substancji. Podróżuje do Wietnamu co sześć do dwunastu miesięcy.
Przegląd systemów
negatywny z wyjątkiem tego, co jest szczegółowo opisane w historii obecnej choroby
badanie okulistyczne
ostrość widzenia z/bez korekcji (Snellen)
- prawe oko (OD): 20/300 (brak poprawy przy otworku)
- lewe oko (OS): 20/60-2+2
ruchliwość/wyrównanie oka
pełne ruchy zewnątrzgałkowe w obu oczach (OU)
ciśnienie wewnątrzgałkowe (IOP): (Tonopen)
- OD: 12 mmHg
- OS: 14 mmHg
źrenice
- OD: 4 mm W ciemności, 3 mm w świetle, brak względnej wady źrenic (RAPD)
- OS: 4 mm W ciemności, 3 mm w świetle, brak RAPD
konfrontacja pola widzenia: (Policz palce)
- od: środkowa Szkocja
- OS: całkowita wada niepłodności
zewnętrzna
normalna po obu stronach
badanie lampy szczelinowej
- powieki/rzęsy: normalna ou
- spojówka/twardówka: czysta i cicha ou
- rogówka: 1+ punktowe nadżerki nabłonkowe, bez osadów rogowych OU
- komora przednia: śladowa komórka i flara i głęboka ou
- Iris: normalna Architektura OU
- soczewka: Wyczyść ou
rozszerzone badanie dna oka (DFE)
- szkliste: śladowe przednie komórki szkliste OU
- dysk:
- OD: Stopień 3 obrzęk tarczy, hiperemiczny
- OS: obrzęk tarczy 2-3 stopnia, hiperemiczny
- stosunek kubka do tarczy: 0,0 ou
- plamka:
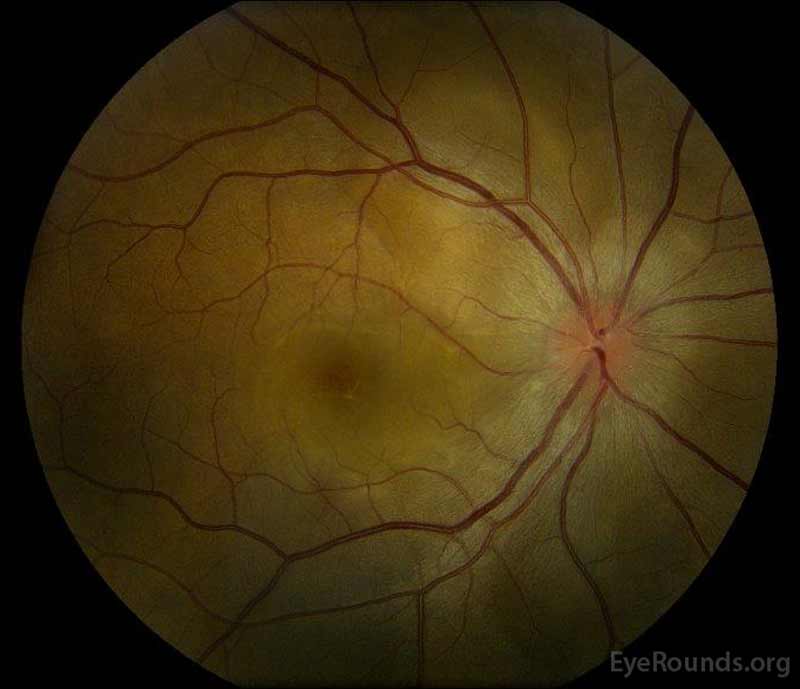
- od: 3+ Cystoid macular edema (CME) i płyn podretinalny (SRF) rozciągający się od tarczy do plamki skroniowej. Brak lipidów i wysięków. Zabagniona naczyniówka.
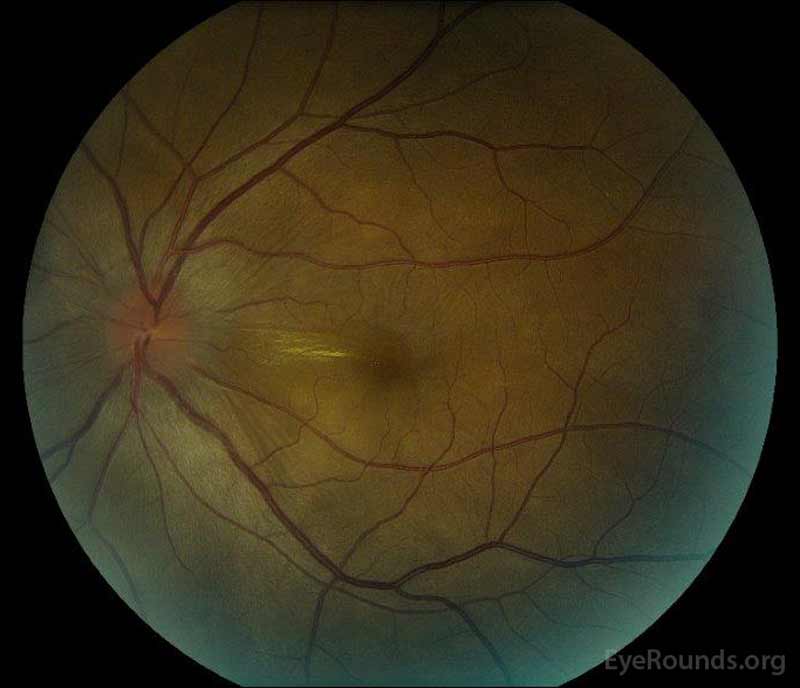
- OS: 2+ CME i SRF rozciągające się od płyty przez fovea. 1-2 + liniowy lipid rozciągający się od dysku w kierunku fovea. Zabagniona naczyniówka.
- naczynia:
- OD: pochewka czasowa
- OS: normalny
- Peryferia:
- OD: torbielowata Kępka siatkówki przed równikiem o 10: 30
- OS: płytkie SRF przed równikiem o 4:00
 |
 |
 |
 |
 |
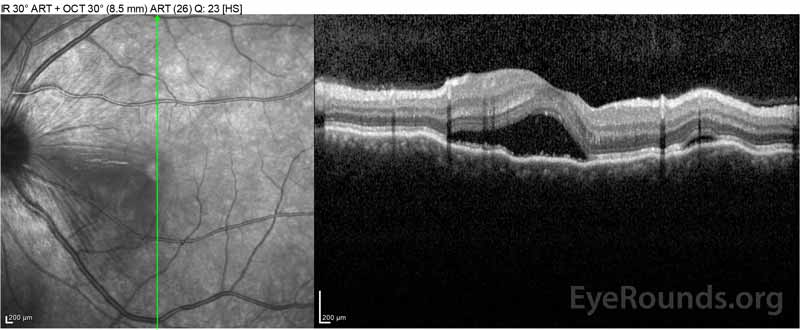 |
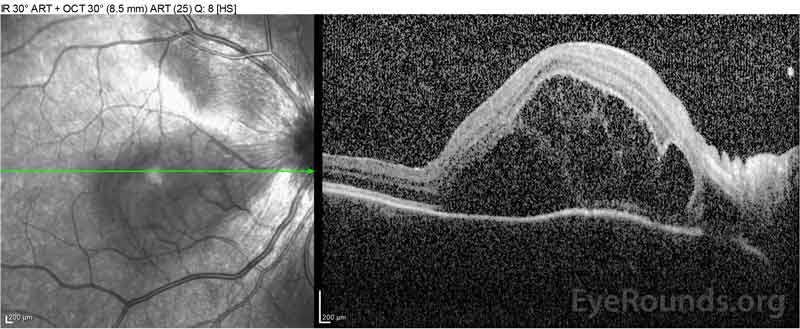
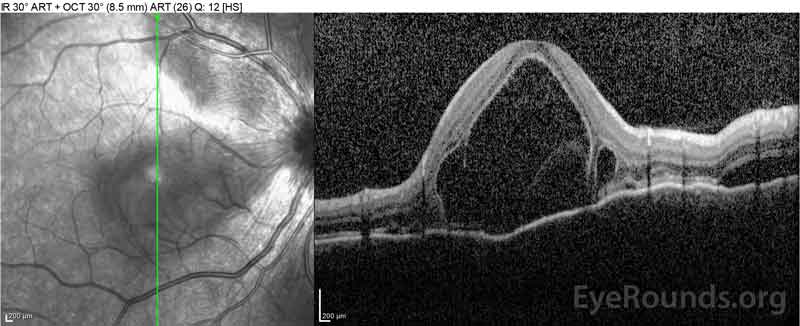
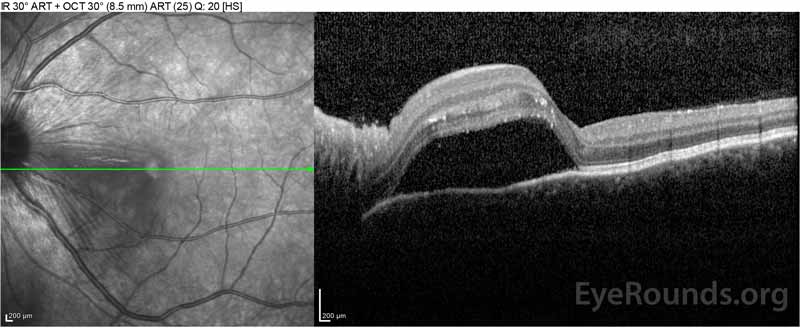
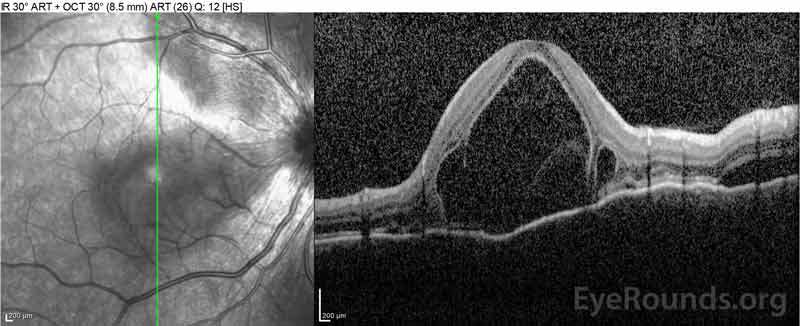
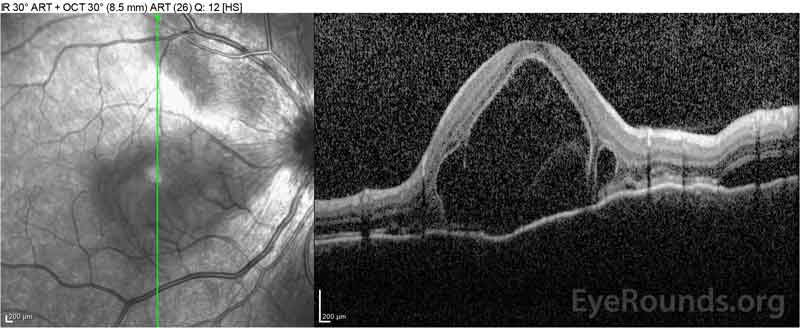
Figure 2: Optyczna tomografia koherentna (OCT) prawego oka (górne panele) pokazuje surowicze odwarstwienie siatkówki z udziałem fovea z rozległym leżącym płynem wewnątrzretinalnym, zakłócenie zewnętrznych warstw siatkówki i pofalowania zagęszczonej naczyniówki. OCT lewego oka (dolne panele) pokazuje surowicze odwarstwienie siatkówki w plamce nosowej rozciągające się aż do fovea.
 |
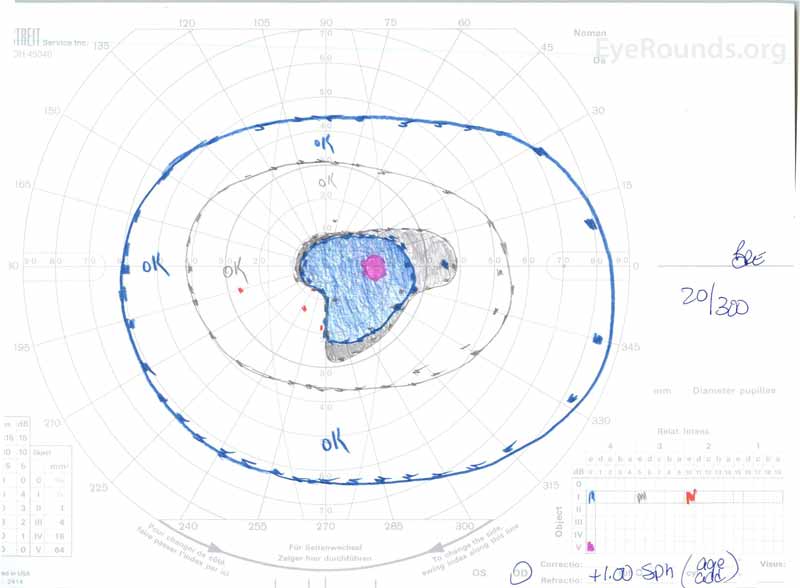 |
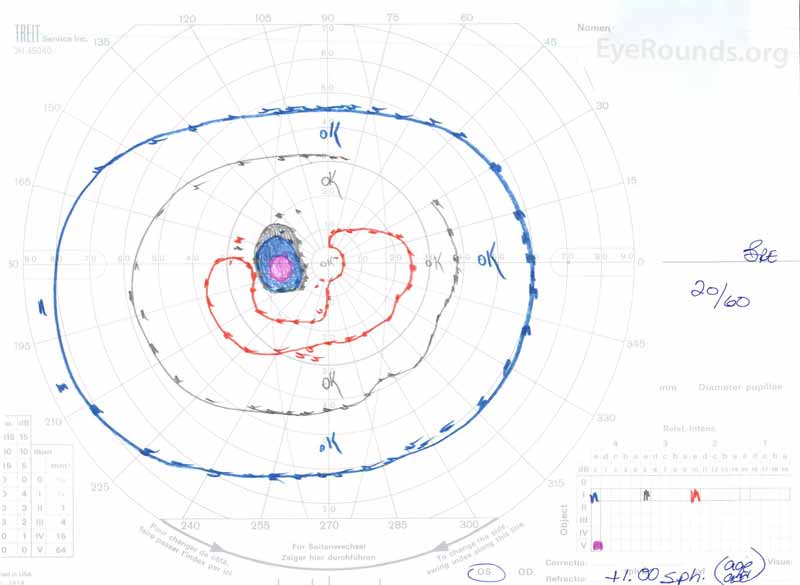
rysunek 3: Goldman visual fields (GVF), OU. (Lewy obraz) OS pokazuje powiększoną fizjologiczną martwą plamę i łagodną środkową mroczkę. (Prawy obraz) OD pokazuje umiarkowanie gęstą środkową mroczkę zawierającą fizjologiczny martwy punkt i rozszerzającą się przedporodowo.
b-scan: brak oznak zapalenia twardówki, łagodne zmętnienie ciała szklistego/komórki inferiorly
diagnostyka różnicowa
- ostra tylna wieloogniskowa nabłonek pigmentowy (APMPPE)
- Centralna surowicza chorioretinopatia
- zapalenie nerwu wzrokowego
- Panuveitis
- choroba autoimmunologiczna (np. SLE, sarkoidoza)
- infekcja (np.G., kiła, gruźlica, Bartonella henselae)
- nowotwór złośliwy (np. chłoniak oczny)
- tylne zapalenie twardziny
- współczulna ophthalmia
- zespół wysięku Uvealnego
- zespół Vogt-Koyanagi-Harada
praca
morfologia krwi
liczba krwinek białych: 4,9 k/mm3 (ref: 3,7-10,5)
liczba krwinek czerwonych 3,99 m/mm3 (ref: 4,0-5,2)
hemoglobina 11,6 g/dl (Ref: 11,9-15,5)
hematokryt 35 % (ref: 35-47)
Basic metabolic panel
Sodium 138 mEq/L (Ref: 135-145)
Potassium 4.3 mEq/L (Ref: 3.5-5.0)
Chloride 107 mEq/L (Ref: 95-107)
CO2 20 mEq/L (Ref: 22-29)
Blood urea nitrogen 16 mEq/dL (Ref: 10-20)
Creatinine 0.7 mg/dL (Ref: 0.5-1.0)
C-reactive Protein (CRP): <0.5 mg/dL (Ref: <=0.5)
Erythrocyte sedimentation rate (ESR): 12 mm/Hr (Ref: 0-20)
Angiotensin–converting enzyme (ACE): 13 U/L (Ref: 8-52)
QuantiFERON-TB Gold: ujemny
żelazo, krew 54 mikrogramy/dL (Ref: 37-145)
całkowita zdolność wiązania żelaza 379 mikrogram/dL (Ref: 250-425)
przebieg kliniczny
pacjentka była początkowo oceniana przez oddział ratunkowy, biorąc pod uwagę jej dolegliwości z powodu nowo pojawiających się ciężkich bólów głowy i utraty wzroku. Tomografia komputerowa mózgu (CT) i rezonans magnetyczny (MRI) skany były nijakie. ESR i CRP mieściły się w granicach normy. Klinika Okulistyczna zbadała ją następnego dnia i wykryła obustronne surowicze odrywki siatkówki i zapalenie błony śluzowej. ACE i QuantiFERON-TB Gold labs były negatywne. Zdiagnozowano u niej chorobę Vogt-Koyanagi-Harada na podstawie jej klinicznej prezentacji i pochodzenia azjatyckiego. Była leczona 80mg prednizonu dziennie, paracetamol w razie potrzeby na bóle głowy i suplementacji witaminy D i wapnia. Jej bóle głowy szybko ustąpiły, a ostrość wzroku stopniowo poprawiała się w ciągu następnych dwóch tygodni. Jej dawka prednizonu zmniejszała się następnie do 40 mg w ciągu trzech tygodni z dalszym ustępowaniem objawów i poprawą ostrości wzroku. Nie miała nawrotów bólów głowy ani pogorszenia widzenia podczas stożka prednizonu. Na ostatniej wizycie zmniejszyła się do 5 mg co drugi dzień, bez powrotu objawów. Jej ostrość wzroku podczas wizyty kontrolnej wynosiła 20/15-2 OD i 20/20+2 OS, a Oct plamki żółtej wykazał pełną rozdzielczość obrzęku dysku i surowiczych odrywek siatkówki w obu oczach(ryc. 4).
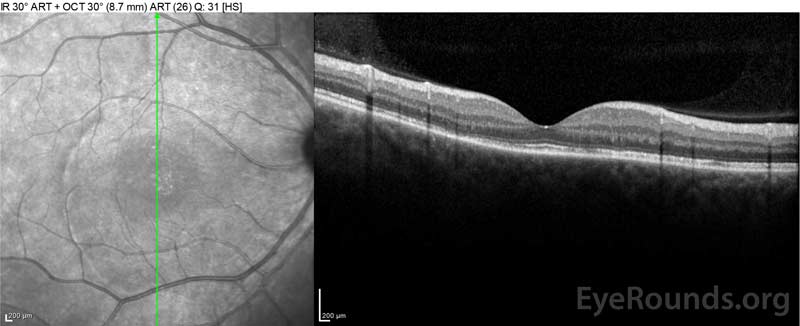
Rysunek 4: Optyczna tomografia koherentna pokazująca płyn podretinalny w punkcie wyjściowym (góra) i przebieg ustąpienia po jednym tygodniu (środek) i pięciu tygodniach (dół), podczas gdy na wysokiej dawce doustnego stożka prednizonu. Zwróć uwagę na wygładzanie fal naczyniówkowych z leczeniem.
 |
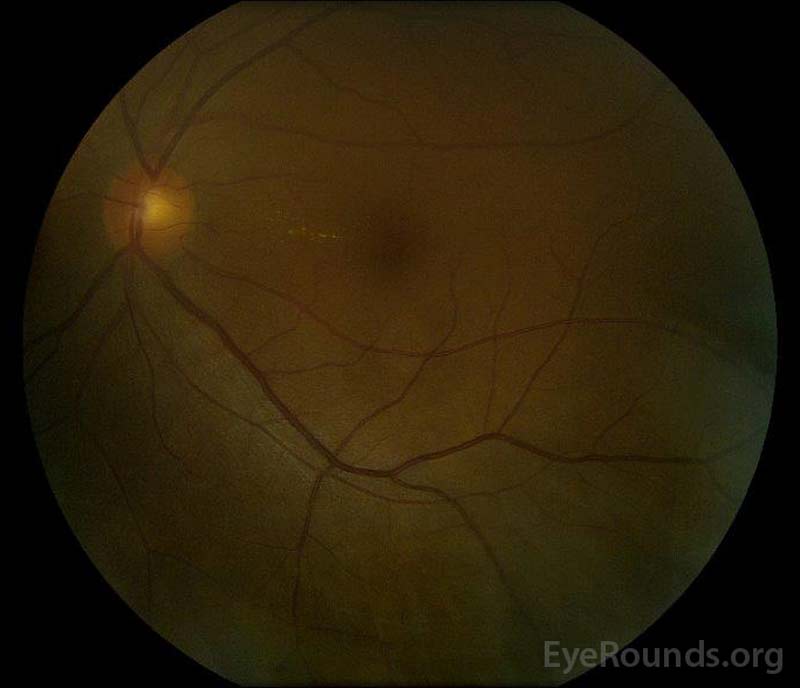 |
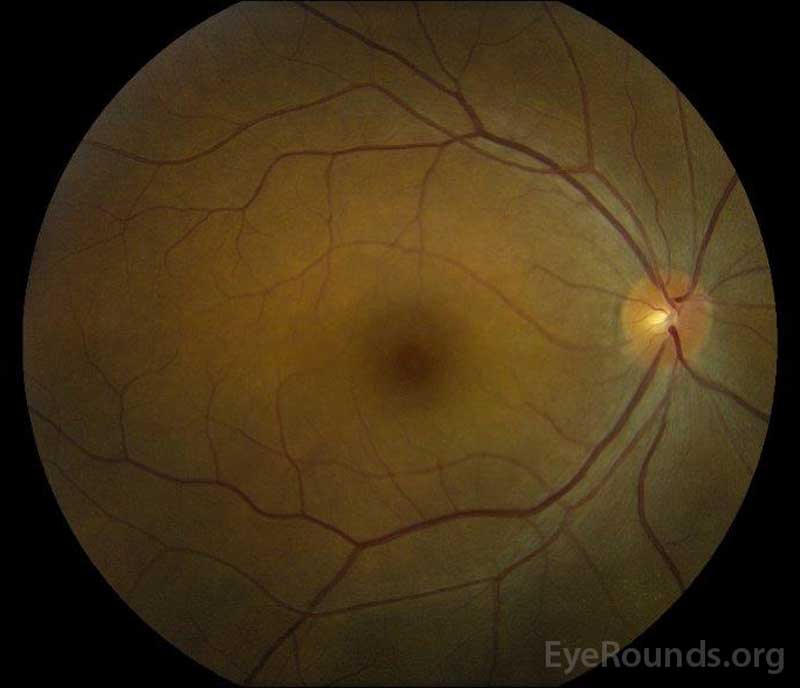
rysunek 5: Kolor dna oka fotografii prawej (a) i lewej (B) oczy podczas fazy rekonwalescencji wykazując poprawę w płynie podretinalnym i obrzęk dysku.
diagnostyka
niekompletna choroba Vogt-Koyanagi-Harada
dyskusja
choroba Vogt-Koyanagi-Harada (VKH) jest ogólnoustrojowym schorzeniem autoimmunologicznym charakteryzującym się obustronnym, nie nekrotyzującym ziarniniakowym zapaleniem panuveitis związanym z pozajelitowymi zmianami w obrębie skóry, takimi jak polioza i bielactwo oraz zapaleniem błony naczyniowej oka, ucha wewnętrznego, włosów i opon mózgowych. Choroba Harada jest izolowanym zapaleniem błony naczyniowej oka bez ogólnoustrojowych objawów VKH.
etiologia
etiologia choroby VKH jest nadal w dużej mierze nieznana pomimo obecnych wysiłków badawczych. Uważa się, że jest to nabyta choroba autoimmunologiczna, polegająca na pośredniczonej przez komórki T nadwrażliwości na melanocytowe antygeny, z podstawową predyspozycją genetyczną i możliwym wyzwalaczem drobnoustrojów . Tyrozynaza i peptydy związane z tyrozynazą są antygenami melanocytów, które zostały zasugerowane jako cele procesów autoimmunologicznych w VKH . Jednak zwiększone ryzyko choroby VKH nie było związane z rodziną genów tyrozynazy, zgodnie z jednym z badań .
ze względu na zwiększoną częstość występowania wśród niektórych grup etnicznych i płciowych uważa się, że istnieje genetyczna predyspozycja do patogenezy VKH. Wiele genów, w tym ludzki antygen leukocytów (HLA) i geny interleukiny (IL), były związane z VKH w różnych populacjach etnicznych . Receptory HLA są głównymi kompleksami zgodności histologicznej u ludzi, które prezentują peptydy do układu odpornościowego. HLA-DR1, HLA-DR4, HLA-DRB1*0405 i HLA-DRw53 to kilka haplotypów występujących u pacjentów z chorobą VKH . HLA-DR4 występuje częściej u Japończyków i Latynosów, natomiast HLA-DRB1*0405 występuje częściej u Koreańczyków i pacjentów z Bliskiego Wschodu . Zarówno allele HLA-DR4, jak i HLA-DRB1*0405 występują u pacjentów wietnamskich . Receptor HLA-DRB1 wiąże się z antygenami melanocytów w różnych pojemnościach. Pomimo tych skojarzeń, badania genetyczne nie są obecnie zalecane.
biorąc pod uwagę zwykłe objawy prodromalne, które towarzyszą VKH, w tym gorączkę, bóle głowy, meningismus i szum w uszach, podżegające etiologia wirusowa została zasugerowana jako wyzwalacz wystąpienia VKH poprzez mechanizmy mimikry molekularnej u genetycznie predysponowanych pacjentów. Koperta cytomegalii glikoproteina h ma znaczącą homologię aminokwasową do peptydu tyrozynazy, a zakażenie CMV może wywołać VKH poprzez mimikrę molekularną (tj. rozpoznawanie przez receptory HLA klasy II). Wirus Ebsteina-bar (EBV) został również zamieszany. Jednak nie było ostatecznych dowodów dotyczących wirusowej etiologii VKH i pozostaje niejasne, co wyzwala odpowiedź autoimmunologiczną VKH .
Patofizjologia
istnieją cztery klasyczne fazy VKH, które mogą mieć zmienne prezentacje: prodromalny, ostry naczynioruchowy, rekonwalescencyjny i przewlekle nawracający. Zmiany histopatologiczne zwykle rozpoczynają się w ostrej fazie .
ostra faza naczyniowa charakteryzuje się obustronnym pogrubieniem błony naczyniowej wtórnym do zapalenia ziarniniakowego. Ziarniniaki składają się z limfocytów, makrofagów i wypełnionych granulkami komórek nabłonkowych i olbrzymich . Chociaż wcześniej uważano, że komórki nabłonkowe są zmienionymi melanocytami ,kontynuacyjne badanie immunohistochemiczne sugerowało pochodzenie z makrofagów tkankowych. Ziarniniaki wypełnione histiocytami nabłonkowymi, nazywane guzkami Dalena-Fuchsa, często można zobaczyć między nabłonkiem barwnikowym siatkówki (RPE) a błoną Brucha. Ziarniniakowe zapalenie błony naczyniowej prowadzi do zgrubienia naczyniówki i wysiękowe odrywki siatkówki wypełnione płynem białkowym. Ponadto pleocytoza (i.e., zwiększona liczba komórek) mogą być obecne w komorze przedniej i ciała szklistego .
Faza rekonwalescencji jest identyfikowana przez depigmentację obszarów naczyniówki i zewnątrzgałkowych, w tym skóry okołogałkowej i włosów. Depigmentowana naczyniówka ustawiona na bladym nerwie wzrokowym sprawia wrażenie” blasku zachodu Słońca ” dna oka, co jest klasyczną cechą tej fazy VKH . Ponadto guzki Dalena-Fuchsa stają się bardziej widoczne pod RPE w fazie rekonwalescencji .
Faza przewlekła-nawracająca charakteryzuje się zmniejszoną grubością naczyniówki, ustępowaniem surowiczych odwarstwień siatkówki, przewlekłym łagodnym zapaleniem witryny i nawracającym zapaleniem ziarniniakowym przedniego odcinka. W tej fazie może rozwinąć się neowaskularyzacja podbarwiona (CNV) i zwłóknienie podbarwione, które są wskaźnikami ciężkiej progresji choroby . Zaćma i jaskra wtórna są innymi powikłaniami długotrwałego lub nawracającego zapalenia w tej fazie .
Epidemiologia
VKH jest rozpowszechniony u ras o ciemniejszym pigmencie skóry, zwłaszcza Azjatów, Ameryki Południowej, Bliskiego Wschodu i rdzennych Amerykanów. Choroba VKH stanowi>10% zapalenia błony naczyniowej oka w tych populacjach . Tylko 1-4% przypadków zapalenia błony naczyniowej oka uważa się za wtórne do choroby VKH w Stanach Zjednoczonych (7). W Stanach Zjednoczonych, większość przypadków VKH okazały się wpływać na osoby z Azji, Hiszpanie, i / lub Native American przyzwoity . Co ciekawe, choroba VKH rzadko dotyka Afrykanów pomimo ich ciemnej pigmentacji . Częstość występowania choroby VKH różni się znacznie między podgrupami rasowymi w sąsiednich krajach . Na przykład częstość występowania VKH w Korei wynosi tylko 2%, znacznie mniej niż w Japonii i Chinach .
VKH ma typowy początek w wieku od 20 do 50 lat ; jednak badania sugerują, że 3,1-13,4% przypadków VKH to pacjenci pediatryczni, a 10% przypadków to ≥65 lat . Klasycznie uważa się, że VKH ma upodobanie do płci żeńskiej i podczas gdy większość badań pokazuje, że VKH nieproporcjonalnie wpływa na kobiety, kilka badań wykazało predyspozycje męskie lub brak predyspozycji do płci .
objawy
jak wspomniano powyżej, cztery etapy choroby VKH są prodromalne, naczyniówki, rekonwalescent, i przewlekłe nawracające. Każdy etap wykazuje różne cechy kliniczne.
- Prodromal: ten początkowy etap może występować jako choroba grypopodobna z głównie objawami konstytucyjnymi, takimi jak ból głowy, zawroty głowy, gorączka, zmęczenie i (lub) nudności. Zgłaszano objawy neurologiczne zapalenia opon mózgowo-rdzeniowych, porażenia nerwu czaszkowego i zapalenia nerwu wzrokowego, a także objawy słuchowe szumu w uszach, dysakuzji i zawrotów głowy . Światłowstręt, niewyraźne widzenie, męty i (lub) ból oka zwykle zaczynają się w ciągu 48 godzin od wystąpienia objawów prodromalnych . Faza prodromalna zwykle trwa od kilku dni do tygodni.
- ostry obrzęk naczynioruchowy: ten etap obejmuje niewyraźne widzenie, światłowstręt, wstrzyknięcie spojówki i ból oka. Nie może być łagodne zapalenie przedniej błony naczyniowej oka, które początkowo wydaje się nie ziarniniakowe. Jednostronny początek zazwyczaj przechodzi w obustronne zaangażowanie w ciągu 1-2 tygodni. Może rozwinąć się ziarniniakowe zapalenie przedniej błony naczyniowej z baranim tłuszczem rogowym. Wyniki badania tylnego mogą obejmować obrzęk nerwu wzrokowego i przekrwienie, wieloogniskowe obszary zapalenia naczyniówki, wiele obszarów surowiczych odrywek siatkówki zlokalizowanych do tylnego dna oka, zgrubienie naczyniówki, promieniujące fałdy chorioretinal, i vitritis . Surowicze odrywki siatkówki mogą tworzyć wzór koniczyny w tylnym dnie i może przejść do rozległych odrywek pęcherzowych w ciężkich przypadkach . Ostra jaskra zapalna była związana z tą fazą choroby i może występować z płytką komorą przednią wtórną do rzęskowego obrzęku ciała, naśladując ostre zamknięcie kątowe . Czas trwania ostrej fazy naczyniówki zależy od szybkiej diagnozy i postępowania.
- przewlekłe naczyniówki lub Rekonwalescent: ten etap zazwyczaj rozwija się kilka tygodni po ostrej fazie i charakteryzuje bielactwo (na przykład, twarz, ręce, ramiona, lub z powrotem), polioza, i łysienie . Depigmentacja w pobliżu limbusu rogówki, znana jako znak Sugiura, może być widoczna miesiąc po rozpoczęciu choroby; jednak ten znak jest rzadko spotykany poza populacją japońską . Depigmentacja naczyniówkowa zwykle występuje w ciągu kilku miesięcy i skutkuje jasnym pomarańczowo-czerwonym kolorem naczyniówki i klasycznym ” sunset glow dna oka.”Sunset glow dna oka jest uważany za najważniejszy i predykcyjny w diagnostyce przewlekłego VKH . Dobrze zaznaczone, okrągłe, nummular chorioretinal blizny mogą tworzyć w środkowej części obwodu. Przewlekła Faza uveitic zazwyczaj trwa kilka miesięcy.
- przewlekłe-nawracające: Etap ten charakteryzuje się nawracającymi epizodami ziarniniakowego zapalenia przedniej błony naczyniowej z baranimi wytrąceniami rogówki, guzkami tęczówki, depigmentacją tęczówki, synechiae tylnymi, tylną zaćmą podkapsułkową, jaskrą wtórną, neowaskularnymi błonami naczyniówkowymi, a ostatecznie zwłóknieniem podretinalnym i atrofią nummular chorioretinal . Faza przewlekła zwykle rozwija się co najmniej sześć miesięcy po wstępnej prezentacji. Surowicze odrywki siatkówki obecne podczas fazy ostrej i rekonwalescencji zwykle nie powtarzają się przy agresywnym leczeniu kortykosteroidami .
kryteria diagnostyczne
najnowsze kryteria diagnostyczne, nazwane Zrewidowanymi kryteriami diagnostycznymi (RDC) dla VKH, zostały zdefiniowane w 1999 r .podczas pierwszych międzynarodowych warsztatów na temat VKH. Przedstawiono je w tabeli 1. RDC są użyteczne w tym, że dzielą VKH na trzy różne kategorie diagnostyczne w oparciu o fazę choroby, podczas której pacjent przedstawia: kompletny, niekompletny i prawdopodobny. Ta Kategoryzacja choroby pozwala na odpowiednie i wczesne leczenie w” prawdopodobnej „chorobie, która może pomóc w zapobieganiu progresji do” kompletnej ” choroby.
prace nad innymi przyczynami zapalenia oka, zarówno zakaźnego, jak i autozapalnego, są niezbędne. Mogą one obejmować szybkość sedymentacji erytrocytów (ESR), białko C-reaktywne (CRP), badanie quantiferon-Gold w kierunku gruźlicy, odczynnik szybkiego osocza (RPR) w przypadku kiły, enzym konwertujący angiotensynę (ACE) i zdjęcie rentgenowskie klatki piersiowej w przypadku sarkoidozy, przeciwciało przeciwjądrowe (ANA) I P-/C-ANCA. Również, historia niedawnego urazu oka lub chirurgii wewnątrzgałkowej musi być odnotowana i prawdopodobnie sugeruje współczulną okulistykę (SO) jako bardziej prawdopodobną diagnozę biorąc pod uwagę bardzo podobną prezentację i patofizjologię dzieloną między SO I VKH .
aby wesprzeć diagnozę VKH w dwuogniskowych przypadkach, można wykonać nakłucie lędźwiowe w celu wykrycia pleocytozy limfocytowej i monocytarnej; jest to jednak rzadko stosowane klinicznie. Osiemdziesiąt procent pacjentów ma pleocytozę w płynie mózgowo-rdzeniowym (CSF) w ciągu jednego tygodnia, a 97% ma pleocytozę w ciągu trzech tygodni. Podwyższony poziom komórek odpornościowych może trwać do ośmiu tygodni po wystąpieniu choroby . Profile markerów powierzchniowych komórek T są podobne w płynie mózgowo-rdzeniowym i cieczy wodnistej, ale różnią się od krwi. Sugeruje to zdolność CSF do dokładnego odzwierciedlenia zapalenia błony naczyniowej w chorobie VKH .
Tabela 1. Zmienione kryteria diagnostyczne choroby Vogt-Koyanagi-Harada
*z tabeli 1 w (15).
” całkowita choroba Vogt-Koyanagi-Harada (kryteria 1 do 5 muszą być obecne)
- brak przeszczepionego urazu ocznego lub operacji chirurgicznej poprzedzającej początkowe wystąpienie zapalenia błony naczyniowej oka.
- brak klinicznych lub laboratoryjnych dowodów wskazujących na inne jednostki chorobowe oka.
- obustronne zaangażowanie oka (a lub b muszą być spełnione, w zależności od stadium choroby, gdy pacjent jest badany).
- wczesne objawy choroby.
- musi istnieć dowód na rozlane zapalenie naczyniówki (z lub bez zapalenia przedniej błony naczyniowej oka, szklistej reakcji zapalnej lub przekrwienia dysku wzrokowego), które może objawiać się jako jeden z następujących objawów:
- wczesne objawy choroby.
- ogniskowe obszary płynu podretinalnego lub
- pęcherzowe surowicze odrywki siatkówki.
- z dwuogniskowymi ustaleniami dna oka; oba z poniższych elementów muszą być również obecne:
- ogniskowe obszary opóźnienia w perfuzji naczyniówkowej, wieloogniskowe obszary punktowego wycieku, duże obszary placoidalne hiperfluorescencji, łączenie się w płynie podretinowym i barwienie nerwu wzrokowego (wymienione w kolejności sekwencyjnej) za pomocą angiografii fluoresceinowej i
- rozproszone zgrubienie naczyniówkowe, bez oznak tylnego zapalenia twardówki za pomocą ultrasonografii.
- Historia sugerująca wcześniejszą obecność wyników z 3A i ZARÓWNO (2), jak i (3) poniżej lub wiele objawów z (3):
- depigmentacja oka (jeden z następujących objawów jest wystarczający): (a) sunset glow dna oka lub (b) znak Sugiura.
- inne objawy oczne:
- zdrętwiałe blizny depigmentowane naczyniowo-siatkówkowe lub
- zlepianie się i/lub migracja nabłonka barwnikowego siatkówki lub
- nawracające lub przewlekłe zapalenie przedniej błony naczyniowej oka.
- Meningismus (złe samopoczucie, gorączka, ból głowy, nudności, ból brzucha, sztywność szyi i pleców lub kombinacja tych czynników; sam ból głowy nie jest jednak wystarczający do spełnienia definicji meningismus), lub
- szum w uszach lub
- pleocytoza płynu mózgowo-rdzeniowego.
- łysienie lub
- Polioza lub
- bielactwo.
niekompletna choroba Vogt-Koyanagi-Harada (kryteria 1 do 3 i muszą być spełnione 4 lub 5)
- brak przenika urazu oka lub operacji chirurgicznych poprzedzających początkowy początek zapalenia błony naczyniowej oka i
- brak klinicznych lub laboratoryjnych dowodów wskazujących na inne jednostki chorobowe oka oraz
- obustronne zajęcie oka.
- wyniki neurologiczne/słuchowe; zgodnie z definicją dla całkowitej choroby Vogt-Koyanagi-Harada powyżej lub
- Wyniki ogólne; zgodnie z definicją dla całkowitej choroby Vogt-Koyanagi-Harada powyżej.
prawdopodobna choroba Vogt-Koyanagi-Harada (izolowana choroba oczu; muszą występować kryteria od 1 do 3)
- brak przeszczepu oka lub operacji chirurgicznej poprzedzającej początkowe wystąpienie zapalenia błony naczyniowej oka.
- brak klinicznych lub laboratoryjnych dowodów wskazujących na inne jednostki chorobowe oka.
- obustronne zaangażowanie narządu wzroku zdefiniowane powyżej dla całkowitej choroby Vogt-Koyanagi-Harada. „
badania / prace laboratoryjne
w początkowym badaniu VKH należy rozważyć uzyskanie następujących testów:
- optyczna tomografia koherencyjna (OCT): W ostrej fazie naczyniówki, OCT będzie prawdopodobnie wykazywać znaczne zgrubienie naczyniówki i surowicze odrywki siatkówki. Subretinal fluid nagromadzenia mogą mieć przegrody uważa się błon fibrynowych i produktów zapalnych, tworząc strukturę lobular które mogą być również postrzegane na fluoresceiny angiografii. W fazie rekonwalescencji OCT może wykryć obszary przerzedzenia siatkówki po ustąpieniu stanu zapalnego po leczeniu kortykosteroidami .
- B-scan ultrasonografia: W ostrej fazie, ultrasonografia może wykazywać rozproszone tylne naczyniówki zgrubienie, tylne twardówki zgrubienie, odłączenie siatkówki, i zmętnienia ciała szklistego . Wysięk rzęskowy można zaobserwować za pomocą biomikroskopii ultradźwiękowej . Test ten jest również przydatny do wykluczenia tylnego zapalenia twardówki.
- angiografia Fluoresceinowa (FA): klasycznie, FA ujawnia wieloogniskowe podfluorescencyjne kropki we wczesnej fazie, a następnie wiele ogniskowych obszarów nadfluorescencyjnych z rozproszonym wyciekiem w późnej fazie . Barwnik przecieka przez RPE i gromadzi się w przestrzeni subretinalnej otaczającej kropki hyperfluorescencyjne. FA może być przydatna diagnostycznie, gdy choroba VKH prezentuje się bez objawów zewnątrzgałkowych. Na śródpiersiu można zaobserwować hiperfluorescencję tarczy nerwu wzrokowego i wady okna spowodowane zanikowymi bliznami chorioretinalnymi . FA w przewlekłym nawracającym stadium choroby VKH wykazuje niespecyficzne wady okna z powodu uszkodzenia RPE, neowaskularyzacji naczyniówkowej i zwłóknienia podretinalnego .
- angiografia zieleni Indocyjaninowej (ICG) : Wczesna faza ICG przedstawia hiperfluorescencyjne naczynia zrębowe, które wskazują na naczyniówkę naczyniówkową i podfluorescencyjne ciemne kropki, które odpowiadają ziarniniakom i opóźnionemu, niejednolitemu wypełnieniu naczyń naczyniówkowych . Późna Faza ujawnia rozmyte zrębowe wzory naczyniowe i rozproszoną hiperfluorescencję choroidalną. Hiperfluorescencja krążka wskazuje na ciężką chorobę. ICGA może wykryć subkliniczne zapalenie naczyniówkowe w bardzo wczesnych stadiach lub nawet po terapii ogólnoustrojowej .
- nakłucie lędźwiowe: pleocytoza w płynie mózgowo-rdzeniowym występuje u większości pacjentów z VKH. Nakłucie lędźwiowe należy wykonać na początku przebiegu choroby, ponieważ pleocytoza może rozwiązać
leczenie/Postępowanie/wytyczne
cele leczenia w VKH obejmują wczesną diagnozę i tłumienie aktywnego zapalenia, wraz z zapobieganiem nawracającym zapaleniom i powikłaniom zagrażającym widzeniu, takim jak jaskra, pęcherzowe odwarstwienie siatkówki i neowaskularyzacja naczyniówkowa.
ogólnoustrojowe leczenie kortykosteroidami jest preferowaną terapią w chorobie VKH, zwłaszcza w ostrym stadium naczyniówki. Wykazano, że droga podania kortykosteroidów (doustnie lub dożylnie) nie wpływa na ostrość wzroku ani nie występuje istotne wizualnie powikłania w leczeniu ostrego VKH . W przypadku ciężkiej choroby sugerowanym protokołem jest dożylne podawanie metyloprednizolonu przez trzy dni, a następnie doustne podawanie dużych dawek prednizonu. W łagodnej i umiarkowanej chorobie, wysoka dawka doustnego prednizonu może być wystarczająca w dawce 1-2 mg / kg / dobę. Dawkę steroidu należy powoli zmniejszać przez około sześć miesięcy, aby zapobiec nawrotom . Agresywne wczesne leczenie, wraz z seryjnymi testami FA wykazującymi zniknięcie wycieku barwnika przez RPE, może pomóc w zapobieganiu dalszemu postępowi choroby, nawrotom i objawom pozamałżeńskim . Miejscowe sterydy i cykloplegics może zmniejszyć komórki w komorze przedniej i ciała szklistego.
iniekcje do ciała szklistego i pod czopem triamcynolonu były stosowane do krótkotrwałej kontroli zapalenia wewnątrzgałkowego w fazie ostrej lub nawracającej; te miejscowe terapie należy rozważyć w przypadku choroby opornej i u pacjentów, którzy słabo tolerują niekorzystne ogólnoustrojowe skutki uboczne sterydów, biorąc pod uwagę Rozszerzony stożek sterydów. Do ciała szklistego zastrzyki anty-VEGF są czasami stosowane do kontroli neowaskularyzacji poduszkowej oraz w przypadkach uporczywych surowiczych odwarstwień siatkówki.
środki oszczędzające steroidy, w tym antymetabolity, inhibitory kalcyneuryny, leki biologiczne, inhibitory TNF-alfa lub środki cytotoksyczne mogą być stosowane w leczeniu VKH i powinny być starannie monitorowane, często w koordynacji z usługą reumatologiczną . Trwają dyskusje na temat stosowania niesteroidowych leków immunosupresyjnych w leczeniu pierwszego rzutu w chorobie VKH. Jednak niedawne badanie nie wykazało różnic w wynikach między wczesnym leczeniem immunomodulującym pierwszego rzutu (IMT) a leczeniem samym prednizonem . Ponadto, terapie immunosupresyjne i biologiczne są kosztowne i wymagają starannej oceny przed leczeniem, a także częstego monitorowania wyników badań krwi w celu oceny poważnych działań niepożądanych.
w fazie przewlekłego nawrotu częste nawroty mogą sugerować oporność na leczenie kortykosteroidami i sugerować potrzebę oszczędzającego steroidy leczenia immunomodulującego . Preferowanym środkiem w przypadku nawrotów opornych na steroidy lub nietolerancji steroidów jest cyklosporyna . Infliksymab, rytuksymab, adalimumab i interferon alfa-2a są lekami biologicznymi stosowanymi również w leczeniu opornego na leczenie zapalenia błony naczyniowej oka w chorobie VKH.
w leczeniu przedniego zapalenia błony naczyniowej oka często związanego z ostrym VKH, należy przepisać miejscowe sterydy (np. octan prednizolonu 1%) i miejscowe cyloplegię (np. cyklopentolan 1% lub atropina 1%) w zależności od stopnia zapalenia komory przedniej.
powikłania oczne są często związane z chorobą VKH. Biorąc pod uwagę wiele etapów i różnych prezentacji, w których pacjent może przedstawić z VKH, leczenie może być opóźnione w wielu przypadkach. W ciężkich postaciach VKH i nawrotów, zapalenie wewnątrzgałkowe może być trudne do opanowania i może prowadzić do uszkodzenia strukturalnego. U ponad 50% pacjentów występują powiązane powikłania, w tym zaćma, jaskra wtórna, neowaskularne błony naczyniowe, włóknienie podretinalne lub ich kombinacja (rycina 6) .
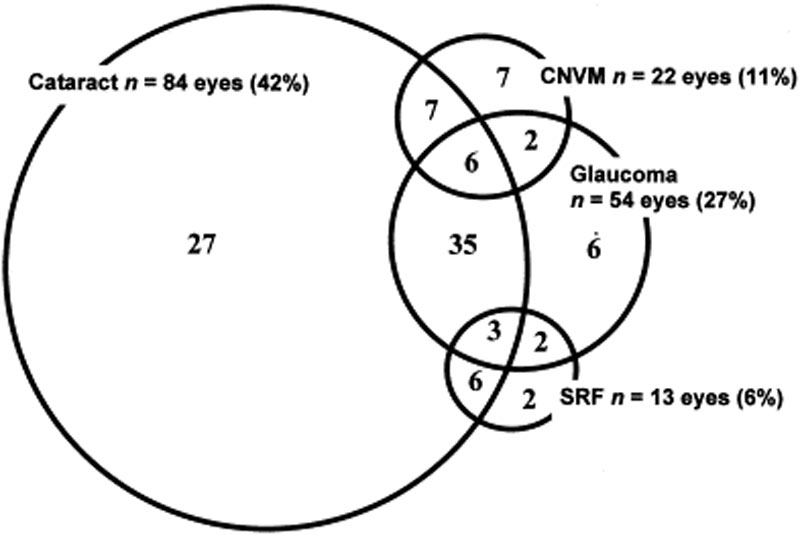
rycina 6: diagram Venna przedstawiający powikłania u pacjentów z VKH. (Wykorzystano za zgodą Am J Ophthalmol. 2001;131(5):599-606 )
epidemiologia i etiologia
|
SIGNS
|
objawy
|
leczenie/postępowanie
|
- Du L, Kijlstra a, Yang P. Vogt-Koyanagi-Harada disease: Novel insights into pathophysiology, diagnosis and treatment. Prog Retin Eye Res 2016; 52: 84-111. https://PubMed.gov/26875727. DOI: 10.1016 / j.preteyeres.2016.02.002
- Yamaki K, Gocho K, Hayakawa K, Kondo I, Sakuragi S. białka z rodziny Tyrozynaz są antygenami specyficznymi dla choroby Vogta-Koyanagi-Harady. J Immunol 2000;165(12): 7323-7329. https://PubMed.gov/11120868
- Horie Y, Takemoto y, Miyazaki a, Namba K, Kase s, Yoshida K, Ota m, Hasumi y, Inoko H, Mizuki N, Ohno S. Rodzina genów tyrozynazy i choroba Vogta-Koyanagi-Harady u pacjentów Japońskich. Mol Vis 2006;12: 1601-1605. https://PubMed.gov/17200659
- Ng JY, Luk FO, Lai TY, Pang CP. Wpływ Genetyki Molekularnej w chorobie Vogt-Koyanagi-Harada. J Ophthalmic Inflamm Infect 2014;4:20. https://PubMed.gov/25097674. DOI: 10.1186 / s12348-014-0020-1
- Bowling B. zapalenie błony naczyniowej oka. Kański ’ s Clinical Ophthalmology New York, New York: Elsevier; 2016; chapter 11; s. 395-465.
- Yeh PT YC, Yang Ch, Lin CP. Niehegmatogenne Odwarstwienie Siatkówki. Na: Schachat AP SS, Hinton DR, Wilkinson CP, Wiedemann P,, editor. Siatkówka Ryana. New York: Elsevier; 2018; chapter 99; s. 1828-1849.
- Goto H RK, Rao N. Vogt–choroba Koyanagi–Harada. In: Schachat AP SS, Hinton DR, Wilkinson CP, Wiedemann P, editor. Siatkówka Ryana. New York, New York: Elsevier; 2018; chapter 78; s. 1505-1515.
- Riddington L, Hall AJ, Tait B, Nicholson i, Varney M. Vogt-Koyanagi-Harada syndrome in patients of Vietnamese rodowód. Aust N Z J Ophthalmol 1996;24(2): 147-149. https://PubMed.gov/9199747
- Sugita S, Takase H, Kawaguchi T, Taguchi C, Mochizuki M. Reakcja krzyżowa peptydów tyrozynazy i antygenu cytomegalii przez komórki T od pacjentów z chorobą Vogt-Koyanagi-Harada. Int Ophthalmol 2007;27 (2-3): 87-95. https://PubMed.gov/17253112. DOI: 10.1007 / s10792-006-9020-y
- Freund BK SD, Mieler WF, Yannuzzi LA. Zapalenie. Atlas Siatkówki. New York, New York: Elsevier 2017; chapter 4; s. 279-398.
- choroba Rao N. Vogt-Koyanagi-Harada. In: J YMaD, editor. Okulistyka. New York, New York: Elsevier; 2014; chapter 7.17; S. 761-763.
- Rao NA, Xu S, Font RL. Oftalmia współczulna. Badanie immunohistochemiczne komórek nabłonkowych i olbrzymich. Okulistyka 1985;92(12):1660-1662. https://PubMed.gov/4088616
- Zespół Vogt-Koyanagi-Harada. In: Whitcup RBNaSM, editor. Zapalenie błony naczyniowej oka: podstawy i praktyka kliniczna. 4th Edition ed: Elsevier; 2010; chapter Chapter 24.
- Read RW, Holland GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L, Pivetti-Pezzi P, Tessler HH, Usui M. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature. Am J Ophthalmol 2001;131(5):647-652. https://PubMed.gov/11336942
- Analiza kliniczna zapalenia błony naczyniowej oka. Korean J Ophthalmol 1989;3(1): 33-37. https://PubMed.gov/2795939. DOI: 10.3341 / kjo1989.3.1.33
- Abu El-Asrar AM, Al-Kharashi AS, Aldibhi H, Al-Fraykh H, kangave D. Vogt-Koyanagi-Harada disease in children. Oko (Lond) 2008;22(9):1124-1131. https://PubMed.gov/17479116. DOI: 10.1038 / sj.oko.6702859
- Martin TD, Rathinam SR, Cunningham ET. Częstość występowania, charakterystyka kliniczna i przyczyny utraty wzroku u dzieci z chorobą Vogt-Koyanagi-Harada w południowych Indiach. Retina 2010; 30 (7): 1113-1121. https://PubMed.gov/20168275. DOI: 10.1097/IAE.0b013e3181c96a87
- Forster DJ, Green RL, Rao NA. Jednostronna manifestacja zespołu Vogt-Koyanagi-Harada u 7-letniego dziecka. Am J Ophthalmol 1991;111(3):380-382. https://PubMed.gov/2000916
- Yamamoto y, Fukushima a, Nishino K, Koura Y, Komatsu T, Ueno H. Vogt-koyanagi-harada choroba z początkiem u pacjentów w podeszłym wieku w wieku od 68 do 89 lat. Jpn J Ophthalmol 2007;51(1):60-63. https://PubMed.gov/17295144. DOI: 10.1007 / s10384-006-0379-0
- Wang Y, Chan CC. Różnice płci w chorobie vogt-koyanagi-harada i oftalmii współczulnej. J Ophthalmol 2014; 2014: 157803. https://PubMed.gov/24734166. DOI: 10.1155/2014/157803
- Nakao K, Abematsu N, Mizushima y, Sakamoto T. obrzęk tarczy nerwu wzrokowego w chorobie Vogt-Koyanagi-Harada. Invest Ophthalmol Vis Sci 2012;53(4):1917-1922. https://PubMed.gov/22408010. DOI: 10.1167/iovs.11-8984
- Rao NA, Gupta a, Dustin L, Chee SP, Okada AA, Khairallah M, Bodaghi B, Lehoang P, Accorinti m, Mochizuki m, Prabriputaloong t, Czytaj RW. Częstość wyróżniania cech klinicznych w chorobie Vogt-Koyanagi-Harada. Ophthalmology 2010;117(3):591-599, 599.e591. https://PubMed.gov/20036008. DOI: 10.1016/j.ophtha.2009.08.030
- Veerappan M, Fleischman D, Ulrich JN, Stinnett SS, Jaffe GJ, Allingham RR. The Relationship of Vogt-Koyanagi-Harada Syndrome to Ocular Hypertension and Glaucoma. Ocul Immunol Inflamm 2017;25(6):748-752. https://PubMed.gov/27438521. DOI: 10.1080/09273948.2016.1189578
- Baltmr A, Lightman S, Tomkins-Netzer O. Vogt-Koyanagi-Harada syndrome – current perspectives. Clin Ophthalmol 2016;10:2345-2361. https://PubMed.gov/27932857. DOI: 10.2147/OPTH.S94866
- Kitaichi N, Matoba H, Ohno S. pozytywna rola nakłucia lędźwiowego w diagnostyce choroby Vogt-Koyanagi-Harada: podgrupy limfocytów w cieczy wodnistej i płynie mózgowo-rdzeniowym. Int Ophthalmol 2007;27 (2-3): 97-103. https://PubMed.gov/17211585. DOI: 10.1007 / s10792-006-9016-7
- Oshima Y, Harino s, Hara Y, Tano Y. indocyanine Green angiographic disease in Vogt-Koyanagi-Harada disease. Am J Ophthalmol 1996;122(1): 58-66. https://PubMed.gov/8659599
- Czytaj RW, Yu F, Accorinti M, Bodaghi B, Chee SP, Fardeau C, Goto H, Holland GN, Kawashima H, Kojima E, Lehoang P, Lemaitre C, Okada AA, Pivetti-Pezzi P, Secchi a, Patrz RF, Tabbara KF, Usui m, Rao NA. Ocena wpływu na wyniki drogi podania kortykosteroidów w ostrej chorobie Vogt-Koyanagi-Harada. Am J Ophthalmol 2006;142(1):119-124. https://PubMed.gov/16815259. DOI: 10.1016 / j.ajo.2006.02.049
- Rubsamen PE, Gass JD. Zespół Vogt-Koyanagi-Harada. Przebieg kliniczny, terapia i długotrwały efekt wizualny. Arch Ophthalmol 1991;109(5):682-687. https://PubMed.gov/2025171
- Urzua CA, Velasquez V, Sabat P, Berger o, Ramirez S, Goecke a, Vásquez DH, Gatica H, Guerrero J. wcześniejsze leczenie immunomodulujące wiąże się z lepszymi efektami wizualnymi u podgrupy pacjentów z chorobą Vogt-Koyanagi-Harada. Acta Ophthalmol 2015; 93 (6):e475-480. https://PubMed.gov/25565265. DOI: 10.1111 / aos.12648
- Czytaj RW, Rechodouni a, Butani N, Johnston R, LaBree LD, Smith RE, Rao NA. Powikłania i czynniki prognostyczne w chorobie Vogt-Koyanagi-Harada. Am J Ophthalmol 2001;131 (5): 599-606. https://PubMed.gov/11336934
sugerowany Format cytowania
Mai AP, Tran C, Wilson CW, Fox AR, Boldt HC. Choroba Vogt-Koyanagi-Harada (VKH). EyeRounds.org. 1 Kwietnia 2019. Dostępne od http://EyeRounds.org/cases/284-vogt-koyanagi-harada.htm
Leave a Reply