Vogt-Koyanagi-Harada (VKH) Ziekte
Auteurs: Anthony P. Mai, BS; Charlene Tran, BS; Caroline W. Wilson, MD; Austin R. Vos, MD; H. Culver Boldt, MD
1 April 2019
INITIËLE PRESENTATIE
hoofdklacht
Wazig zien en hoofdpijn
de Geschiedenis van de Huidige Ziekte
Een 44-jarige Vietnamese vrouw gepresenteerd op de afdeling spoedeisende hulp met een 10-daagse geschiedenis van de progressieve wazig zicht in beide ogen en een drie-daagse voorgeschiedenis van ernstige hoofdpijn. Haar centrale gezichtsverlies was niet verbeterd met een breking door haar optometrist. Haar ernstige occipitale hoofdpijn verergerde met beweging en werden geassocieerd met algemene malaise, extreme vermoeidheid, lichte fotofobie en scheuren. Paracetamol verlichtte gedeeltelijk de pijn.
ze was onlangs naar Vietnam gereisd, maar ontkende dat ze daar zieke contacten had ontmoet. Ze ontkende kaakklaudicatie, koorts of gewichtsveranderingen. Ze ontkende huiduitslag, gehoorveranderingen, tinnitus, duizeligheid, gevoelloosheid of tintelingen. Ze ontkende ooit tuberculose te hebben gehad. Ze had geen voorgeschiedenis van eerdere gezichtsproblemen, auto-immuunziekten of kanker.
oculaire geschiedenis
- voorgeschiedenis van cosmetische ooglidchirurgie (bilaterale blefaroplastie) drie jaar daarvoor
- geen voorgeschiedenis van oculaire trauma of ziekte
medische geschiedenis
geen
geneesmiddelen
Paracetamol indien nodig
allergieën
geen bekende geneesmiddelallergieën
/p>
familiegeschiedenis
geen voorgeschiedenis van oogziekte of auto-immuunziekte
sociale geschiedenis
zij emigreerde enkele jaren voor de presentatie vanuit Vietnam. Ze is getrouwd en heeft drie kinderen. Ze werkt in een nagelsalon. Ze gebruikt geen tabaksproducten, alcohol of illegale stoffen. Ze reist om de zes tot twaalf maanden naar Vietnam.
Evaluatie van Systemen
Negatieve uitzondering van wat is beschreven in de geschiedenis van de huidige ziekte
OCULAIRE EXAMEN
Gezichtsscherpte met/zonder correctie (Snellen)
- Rechter oog (OD): 20/300 (geen verbetering met pinhole)
- Linker oog (OS): 20/60-2+2 (geen verbetering met pinhole)
Oculaire Motiliteit/Uitlijning
Volledige extraocular bewegingen in beide ogen (OU)
de Intraoculaire Druk (IOP): (Tonopen)
- OD: 12 mmHg
- OS: 14 mmHg
Leerlingen
- OD: 4 mm in een donkere, 3 mm in het licht, geen relatieve afferente pupil defect (RAPD)
- OS: 4 mm in een donkere, 3 mm in het licht, geen RAPD
Confrontatie visuele velden: (Aantal vingers)
- OD: Centrale scotoma
- OS: Totaal inferotemporal defect
Extern
Normaal aan beide zijden
spleetlamp examen
- Deksels/wimpers: Normale OU
- Bindvlies/sclera: Duidelijke en rustige OU
- Hoornvlies: 1+ punctate epitheliale erosies, geen keratic mislukt OU
- Anterior chamber: Trace cel en flare en diepe OU
- Iris: de Normale architectuur OU
- Lens: Helder OU
Verwijde fundus onderzoek (DFE)
- Glasvocht: Trace anterior glasvocht cellen OU
- Schijf:
- OD: Leerjaar 3 schijf oedeem, hyperemic
- OS: Leerjaar 2-3 schijf oedeem, hyperemic
- Kop-to-disc ratio: 0.0 OU
- Macula:
- OD: 3+ intestinalis macula-oedeem (CME) en subretinale vloeistof (SRF) uitbreiding van de schijf naar de stoffelijke macula. Geen lipiden of exsudaten. Moerassig-verschijnende choroid.
- OS: 2 + CME en SRF die van de schijf door de fovea lopen. 1-2 + lineair lipide dat zich uitstrekt van schijf naar fovea. Moerassig-verschijnende choroid.
- Schepen:
- OD: temporaal omhulsel
- OS: normaal
- periferie:
- OD: cystische retinale tuft anterior to the equator at 10:30
- OS: ondiepe SRF anterior to the equator at 4:00
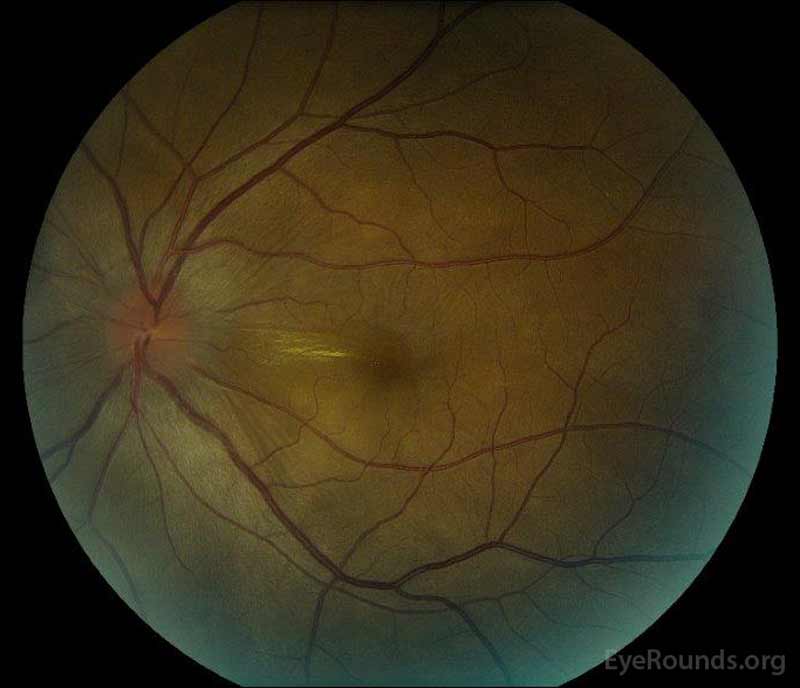
|
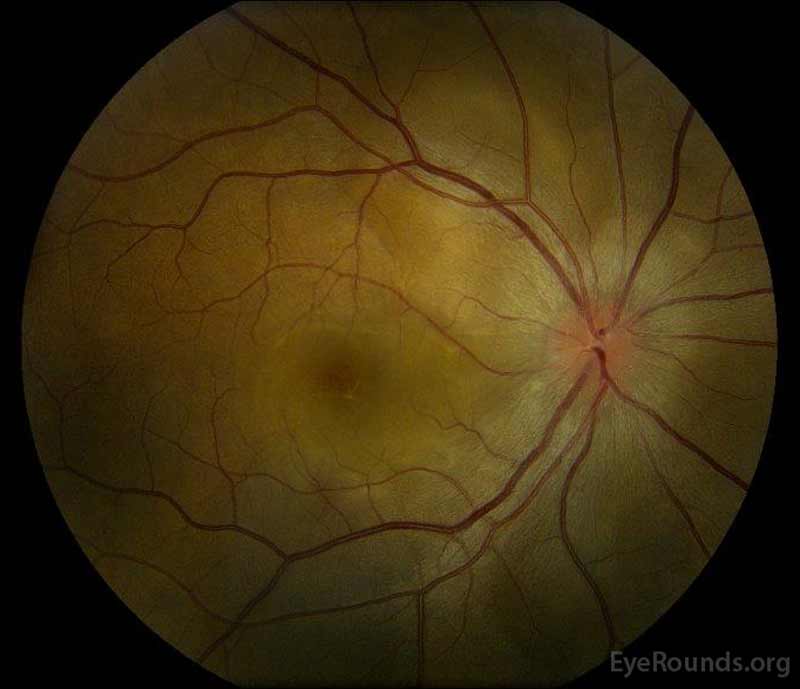
figuur 1: kleurenfoto ‘ s bij presentatie: (afbeelding links) het rechteroog heeft een oedeem van de schijf en milde hyperemie, evenals subretinale vloeistof die zich tijdelijk van de schijf door de macula uitstrekt. Er is ook een focale sereuze netvliesloslating superotemporeel aan de schijf, langs de superieure arcade. (Right image) The left eye has disc edema and mild hyperemia, along with subretinal fluid extending from the disc to the macula and linear lipid deposits in the nasal macula.
 |
 |
 |
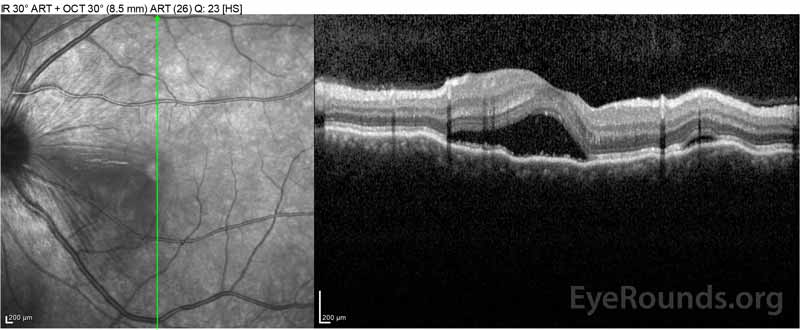 |
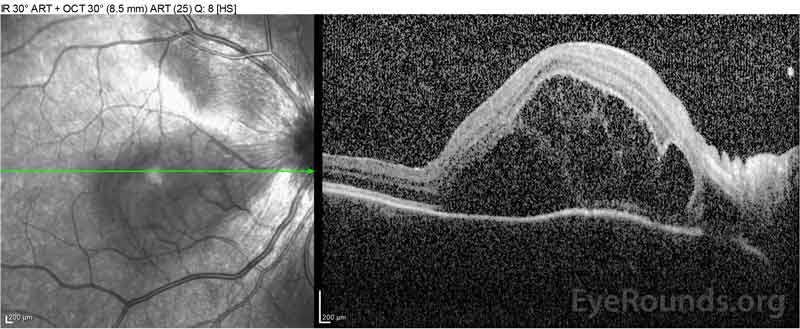
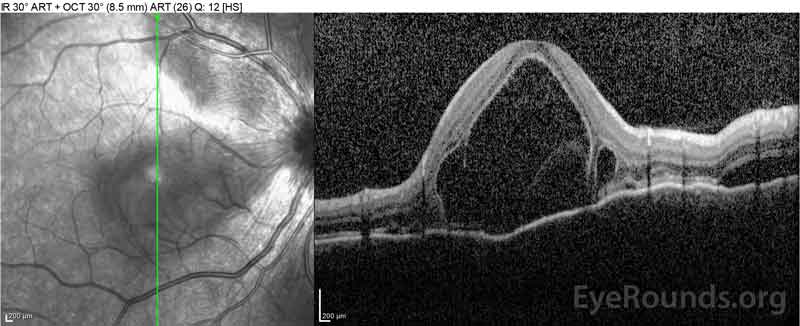
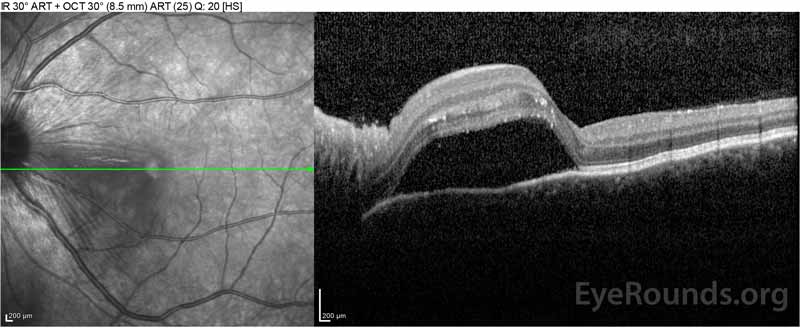
Figure 2: Optical coherence tomography (OCT) van het rechteroog (bovenste panelen) toont een sereuze netvliesloslating waarbij de fovea met uitgebreide bovenliggende intraretinale vloeistof, verstoring van de buitenste retinale lagen, en golvingen van de verdikte choroid. OCT van het linkeroog (onderste panelen) toont een sereuze netvliesloslating in de nasale macula die zich uitstrekt tot aan de fovea.
 |
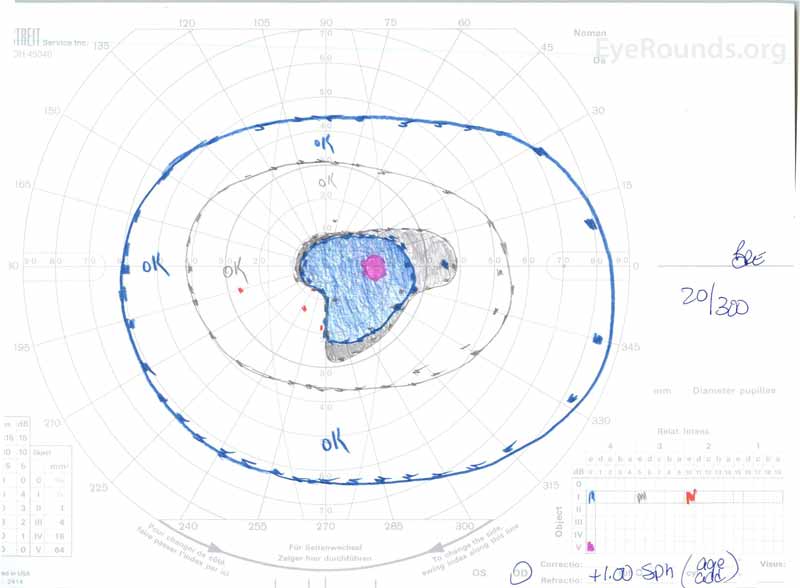 |
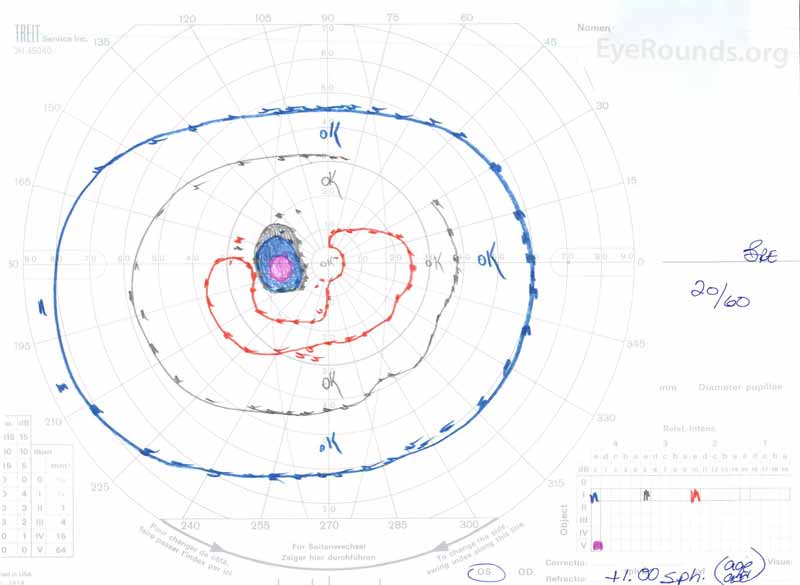
Figuur 3: Goldman visual fields (GVF), OU. (Linker afbeelding) OS toont een vergrote fysiologische blinde vlek en een mild centraal scotoom. OD toont een matig dichte centrale scotoma met daarin de fysiologische blinde vlek en die zich onvruchtbaar uitbreidt.
B-scan: geen tekenen van scleritis, lichte vitreale opaciteiten/cellen inferiorly
differentiële diagnose
- Acute posterieure multifocale placoïde pigmentepitheliopathie (APMPPE)
- centrale sereuze chorioretinopathie
- optische neuritis
- Panuveïtis
- auto-immuunziekte (bijv. SLE, sarcoïdose)
- infectie (e.g., syfilis, tuberculose, Bartonella henselae)
- Maligniteit (bijv., oculaire lymfoom)
- Posterieure scleritis
- Sympathieke oogontsteking
- Uveal effusie syndroom
- Vogt-Koyanagi-Harada Syndroom
onderzoek
Volledig bloedbeeld
Witte bloedcellen: 4.9 K/mm3 (Ref: 3.7-10.5)
Rode bloedcellen 3.99 M/mm3 (Ref: 4.0-5.2)
Hemoglobine van 11,6 g/dL (Ref: 11.9-15.5)
Hematocriet 35 % (Ref: 35-47)
Basic metabolic panel
Sodium 138 mEq/L (Ref: 135-145)
Potassium 4.3 mEq/L (Ref: 3.5-5.0)
Chloride 107 mEq/L (Ref: 95-107)
CO2 20 mEq/L (Ref: 22-29)
Blood urea nitrogen 16 mEq/dL (Ref: 10-20)
Creatinine 0.7 mg/dL (Ref: 0.5-1.0)
C-reactive Protein (CRP): <0.5 mg/dL (Ref: <=0.5)
Erythrocyte sedimentation rate (ESR): 12 mm/Hr (Ref: 0-20)
Angiotensin–converting enzyme (ACE): 13 U/L (Ref: 8-52)
QuantiFERON-TB goud: negatief
ijzer, bloed 54 microgram/dL (Ref: 37-145)
Totale ijzerbindingscapaciteit 379 microgram/dL (Ref: 250-425)
klinisch verloop
de patiënt werd in eerste instantie geëvalueerd door de afdeling spoedeisende hulp, gezien haar klachten van nieuwe ernstige hoofdpijn en verlies van gezichtsvermogen. Hersentomografie (CT) en magnetic resonance imaging (MRI) scans waren onopvallend. ESR en CRP lagen binnen normale niveaus. De oogheelkunde kliniek evalueerde haar de volgende dag en vond bilaterale sereuze netvliesloslating en panuveïtis. ACE en QuantiFERON-TB Gold labs waren beide negatief. Ze werd gediagnosticeerd met de ziekte van Vogt-Koyanagi-Harada op basis van haar klinische presentatie en Aziatische afkomst. Ze werd behandeld met 80 mg prednison per dag, paracetamol als dat nodig is voor hoofdpijn, en vitamine D en calcium suppletie. Haar hoofdpijnen verdwenen snel, en haar gezichtsscherpte verbeterde gestaag gedurende de volgende twee weken. Haar prednison dosering werd vervolgens verminderd tot 40 mg gedurende drie weken met voortdurende vermindering van de symptomen en verbetering van gezichtsscherpte. Ze had geen herhaling van hoofdpijn of verslechtering van het gezichtsvermogen tijdens het afbouwen van de prednison. Op haar meest recente afspraak, ze was gedaald tot 5 mg om de andere dag, zonder terugkeer van de symptomen. Haar gezichtsscherpte bij dat follow-up bezoek was 20/15-2 OD en 20/20+2 OS, en macula OCT toonde volledige resolutie van disc oedeem en sereuze netvliesloslating in beide ogen (Figuur 4).
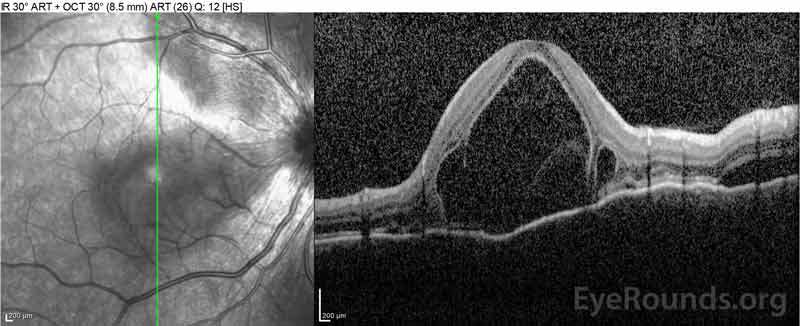
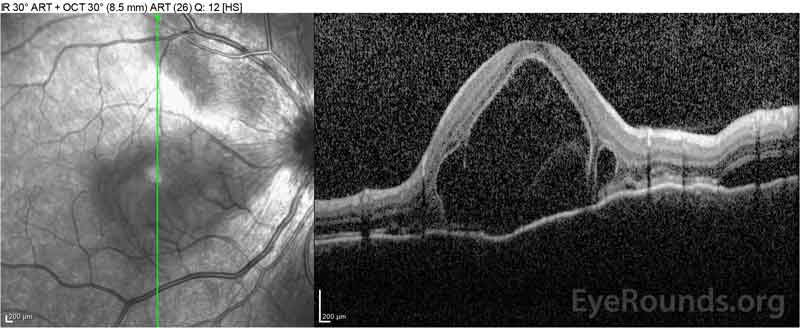
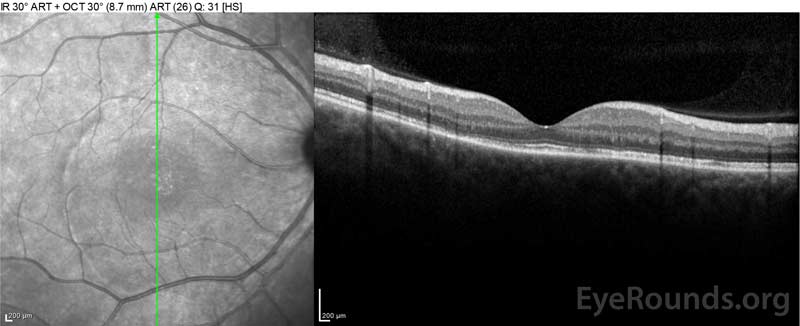
Figuur 4: Optische coherentietomografie met subretinale vloeistof bij baseline (boven) en het verloop van het verdwijnen na één week (Midden) en vijf weken (onder) tijdens een hoge dosis oraal prednison taper. Let op het gladstrijken van de choroïdale golvingen met behandeling.
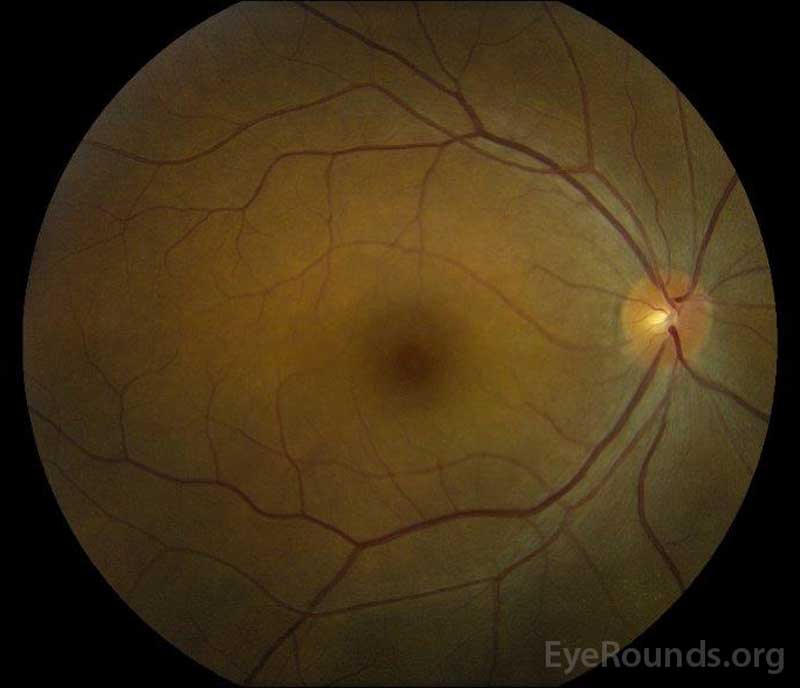 |
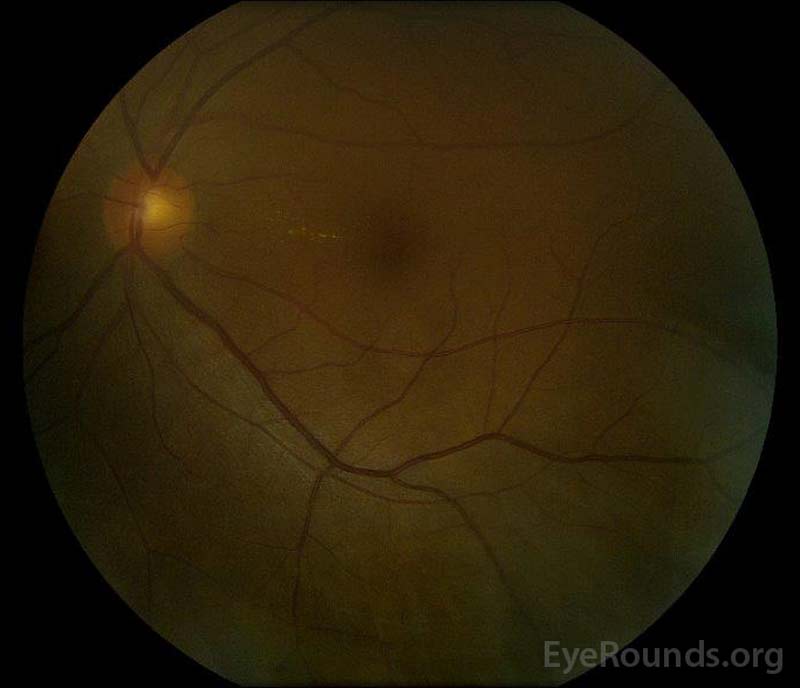 |
Figuur 5: Kleur fundus fotografie van de rechter (A) en linker (B) ogen tijdens de herstelfase aantonen verbetering in de subretinale vloeistof en schijf oedeem.
diagnose
onvolledige ziekte van Vogt-Koyanagi-Harada
discussie
de ziekte van Vogt-Koyanagi-Harada (VKH) is een systemische auto-immuunziekte die wordt gekarakteriseerd door bilaterale niet-necrotiserende granulomateuze panuveïtis geassocieerd met extraoculaire integumentaire veranderingen, zoals poliosis en vitiligo, en ontsteking van de uvea, binnenoor, haar en hersenvliezen. De ziekte van Harada is de geïsoleerde uveïtis zonder de systemische tekenen of symptomen van VKH.
etiologie
de etiologie van VKH-ziekte is ondanks de huidige onderzoeksinspanningen nog grotendeels onbekend. Het wordt verondersteld om een verworven auto-immune ziekte met T-cel-gemedieerde overgevoeligheid voor melanocytic zelf-antigenen te zijn, met een onderliggende genetische predispositie en mogelijke microbiële trigger . Tyrosinase en tyrosinase-verwante peptides zijn melanocyt-antigenen die als doelstellingen van auto-immune processen in VKH zijn voorgesteld . Echter, verhoogd risico op VKH-ziekte was niet geassocieerd met de tyrosinasegenenfamilie, volgens één studie .
vanwege de verhoogde prevalentie onder bepaalde etnische en geslachtsgroepen, wordt aangenomen dat er een genetische predispositie is in de pathogenese van VKH. Meerdere genen, waaronder humaan leukocytenantigeen (HLA) en interleukine (IL) genen, zijn in verband gebracht met VKH in verschillende etnische populaties . HLA-receptoren zijn belangrijke histocompatibiliteitscomplexen bij mensen die peptiden aan het immuunsysteem presenteren. HLA-DR1, HLA-DR4, HLA-DRB1*0405 en HLA-DRw53 zijn verschillende haplotypes gevonden bij patiënten met de ziekte van VKH . HLA-DR4 komt vaker voor bij Japanse en Spaanse mensen, terwijl HLA-DRB1 * 0405 vaker voorkomt bij koreaanse en Midden-Oosterse patiënten . Zowel de HLA-DR4 als de HLA-DRB1*0405 allelen worden gevonden bij Vietnamese patiënten . De HLA-DRB1 receptor bindt aan melanocyt antigenen in verschillende capaciteiten. Ondanks deze associaties wordt genetische tests op dit moment niet aanbevolen.
gezien de gebruikelijke prodromale symptomen die gepaard gaan met VKH, waaronder koorts, hoofdpijn, meningisme en tinnitus, is een opzwepende virale etiologie gesuggereerd als trigger voor het ontstaan van VKH door mechanismen van moleculaire nabootsing bij genetisch gepredisponeerde patiënten. Cytomegalovirus envelop glycoproteïne H heeft significante aminozuurhomologie aan tyrosinasepeptide, en CMV besmetting kan VKH door moleculaire mimicry teweegbrengen (d.w.z., erkenning door HLA-klasse II receptoren) . Ebstein-bar virus (EBV) is ook betrokken. Nochtans, is er geen definitief bewijsmateriaal betreffende een virale etiologie van VKH geweest, en het blijft onduidelijk wat de auto-immune reactie van VKH teweegbrengt .
Pathofysiologie
Er zijn vier klassieke fasen van VKH die variabele presentaties kunnen hebben: prodromal, acuut uveitisch, herstel en chronisch-recidiverend. Histopathologische veranderingen beginnen meestal in de acute fase .
de acute uveitische fase wordt gekenmerkt door bilaterale uveale verdikking secundair aan granulomateuze ontsteking. De granulomen bestaan uit lymfocyten, macrofagen, en korrel gevulde epithelioïde en reuzencellen . Hoewel de epithelioid cellen eerder om veranderde melanocytes werden verondersteld, stelde een follow-up immunohistochemical studie een oorsprong van weefselmacrophages in plaats daarvan voor . Granulomen gevuld met epithelioïde histiocyten, genoemd Dalen-Fuchs knobbeltjes, kunnen vaak worden gezien tussen het retinale pigment epitheel (RPE) en Bruch ‘ s membraan. De uveale granulomateuze ontsteking leidt tot choroïdale verdikking en exsudatieve netvliesloslating gevuld met eiwitvloeistof. Bovendien, pleocytose (i.e., verhoogd aantal cellen) aanwezig kunnen zijn in de voorste oogkamer en glasvocht .
de herstelfase wordt geïdentificeerd door depigmentatie van de choroïde en extraoculaire gebieden, waaronder de perioculaire huid en haar. Een depigmented choroid ingesteld tegen een bleke oogzenuw geeft de indruk van een “sunset-glow” fundus, dat is een klassiek kenmerk van deze fase van VKH . Bovendien worden de knobbeltjes van Dalen-Fuchs steeds prominenter Onder het RPE in de herstelfase .
de chronische recidiverende fase wordt gekenmerkt door verminderde choroïdale dikte, resolutie van sereuze netvliesloslating, chronische lichte vitritis en recidiverende granulomateuze ontsteking van het voorste segment. Choroïdale neovascularisatie (CNV) en subretinale fibrose kunnen zich tijdens deze fase ontwikkelen en zijn indicatoren van ernstige ziekteprogressie . Staar en secundair glaucoom zijn andere complicaties van langdurige of terugkerende ontsteking in deze fase .
Epidemiologie
VKH komt voor bij rassen met een donkerder huidpigment, vooral Aziaten, Zuid-Amerikanen, Midden-Oosteners en Indianen. VKH-ziekte is verantwoordelijk voor >10% van de uveïtis in deze populaties . Slechts 1-4% van uveïtis gevallen worden verondersteld secundair aan VKH ziekte in de Verenigde Staten te zijn (7). In de Verenigde Staten, zijn de meeste gevallen van VKH gevonden om individuen van Aziatische, Hispanic, en/of Inheemse Amerikaanse fatsoenlijk te beà nvloeden . Interessant is dat de ziekte van VKH zelden Afrikanen treft ondanks hun donkere pigmentatie . De incidentie van VKH-ziekte varieert sterk tussen raciale subgroepen in buurlanden . Korea ‘ s incidentie van VKH is bijvoorbeeld slechts 2%, veel lager dan die in Japan en China .
VKH heeft een typisch begin van de leeftijd van 20 tot 50 jaar; studies suggereren echter dat 3,1-13,4% van de gevallen van VKH pediatrische patiënten zijn en dat 10% van de gevallen ≥65 jaar oud is . Klassiek, VKH wordt verondersteld om een voorliefde voor het vrouwelijke geslacht te hebben, en terwijl de meeste studies tonen aan dat VKH onevenredig vrouwen treft, hebben een paar studies een mannelijke aanleg of geen geslacht aanleg aangetoond .
tekenen / symptomen
zoals hierboven vermeld, zijn de vier stadia van de ziekte van VKH prodromaal, uveitisch, herstellend en chronisch recidiverend. Elke fase vertoont verschillende klinische kenmerken.
- Prodromal: deze eerste fase kan zich voordoen als een griepachtige ziekte met voornamelijk constitutionele symptomen, zoals hoofdpijn, duizeligheid, koorts, vermoeidheid en/of misselijkheid. Neurologische symptomen van meningitis, hersenzenuwverlamming en optische neuritis, evenals auditieve symptomen van tinnitus, dysacusis en vertigo zijn gemeld . Fotofobie, wazig zien, drijvers en/of oogpijn beginnen meestal binnen 48 uur na prodromal symptomen . De prodromale fase duurt meestal van een paar dagen tot weken.
- acuut Uveitisch: dit stadium omvat wazig zien, fotofobie, conjunctivale injectie en oogpijn. Er kan milde voorafgaande uveïtis zijn die in eerste instantie niet-granulomateus lijkt. Unilaterale aanvang overgangen naar bilaterale betrokkenheid binnen 1-2 weken. Granulomateuze voorafgaande uveïtis met keratische precipitaten van schapenvet kan zich ontwikkelen. De bevindingen van het posterieure onderzoek kunnen oogzenuwoedeem en hyperemie omvatten, multifocale gebieden van choroiditis, meerdere gebieden van sereuze netvliesloslating gelokaliseerd aan de posterieure fundus, choroïdale verdikking, uitstralende chorioretinale plooien, en vitritis . Sereuze netvliesloslating kan een klaverbladpatroon vormen in de achterste fundus en kan in ernstige gevallen overgaan tot uitgebreide bulleuze detachementen . Acute inflammatoire glaucoom is geassocieerd met deze fase van de ziekte en kan presenteren met een ondiepe voorste kamer secundair aan ciliaire lichaamsoedeem, nabootsen acuut-hoek sluiting . De duur van de acute uveitische fase hangt af van de snelle diagnose en behandeling.
- chronisch Uveitisch of herstellend: dit stadium ontwikkelt zich meestal enkele weken na de acute fase en wordt gekenmerkt door vitiligo (bijv. gezicht, handen, schouders of rug), poliose en alopecia . Depigmentatie in de buurt van het hoornvlies limbus, bekend als Sugiura ‘ s teken, kan worden gezien een maand na het begin van de ziekte ; echter, dit teken wordt zelden gezien buiten de Japanse bevolking . Choroïdale depigmentatie treedt meestal op over een paar maanden en resulteert in de heldere oranje-rode kleur van het choroid en de klassieke “sunset glow fundus.”Sunset glow fundus wordt beschouwd als de belangrijkste en voorspellende in de diagnose van chronische VKH . Goed gedefinieerde, ronde, nummulaire chorioretinale littekens kunnen zich in het midden van de periferie vormen. De chronische uveitische fase duurt meestal enkele maanden.
- chronisch-recidiverend: Dit stadium wordt gekenmerkt door terugkerende episoden van granulomateuze voorafgaande uveïtis met keratische precipitaten van schapenvet, irisnodules, irispigmentatie, posterieure synechiae, posterieure subcapsulaire cataract, secundair glaucoom, choroïdale neovasculaire membranen, en uiteindelijk subretinale fibrose en nummulaire chorioretinale atrofie . De chronische fase ontwikkelt zich meestal ten minste zes maanden na de eerste presentatie. De sereuze netvliesloslating aanwezig tijdens de acute en herstellende fasen meestal niet terugkeren met agressieve corticosteroïde behandeling .
diagnostische Criteria
De meest recente diagnostische criteria, genaamd De herziene diagnostische Criteria (RDC) voor VKH, werden in 1999 gedefinieerd tijdens de eerste internationale Workshop over VKH . Deze zijn weergegeven in Tabel 1. De RDC zijn nuttig omdat ze VKH in drie verschillende diagnostische categorieën verdelen op basis van de ziektefase waarin een patiënt zich presenteert: compleet, onvolledig en waarschijnlijk. Deze categorisering van de ziekte zorgt voor een geschikte en vroege behandeling in “waarschijnlijke” ziekte die kan helpen bij het voorkomen van progressie naar” volledige ” ziekte.
onderzoek naar andere oorzaken van oculaire ontsteking, zowel infectieus als auto-inflammatoir, is essentieel. Deze kunnen de bezinkingssnelheid van de erytrocyten (ESR), C-reactieve proteã ne (CRP), quantiferon-goud testen voor tuberculose, snelle plasmareazin (RPR) voor syfilis, angiotensin-converting enzyme (ACE) en een borst x-ray voor sarcoïdose, antinuclear antilichaam (ANA), en p-/c-ANCA omvatten. Ook moet een geschiedenis van recente oculaire trauma of intraoculaire chirurgie worden opgemerkt en waarschijnlijk suggereert sympathische oftalmie (zo) als de meest waarschijnlijke diagnose gezien de zeer vergelijkbare presentatie en pathofysiologie gedeeld tussen SO en VKH .
om een diagnose van VKH in twijfelachtige gevallen te ondersteunen, kan een lumbaalpunctie worden uitgevoerd om te zoeken naar lymfocytische en monocytische pleocytose; dit wordt echter zelden klinisch toegepast. Tachtig procent van de patiënten heeft binnen één week pleocytose in het cerebrospinale vocht (CSF) en 97% heeft binnen drie weken pleocytose. Verhoogde niveaus van immuuncellen kan duren tot acht weken na het begin van de ziekte . De T-cel oppervlakte marker profielen zijn vergelijkbaar tussen de CSF en het kamerwater maar verschillend van het bloed. Dit suggereert het vermogen van CSF om nauwkeurig uveale ontsteking bij de ziekte van VKH weer te geven .
Tabel 1. Herziene diagnostische Criteria voor de ziekte van Vogt-Koyanagi-Harada
*uit Tabel 1 in (15).
“volledige ziekte van Vogt-Koyanagi-Harada (criteria 1 tot 5 moeten aanwezig zijn)
- geen voorgeschiedenis van penetrerend oculair trauma of chirurgie voorafgaand aan het eerste begin van uveïtis.
- geen klinische of laboratoriumgegevens die wijzen op andere entiteiten met oculaire aandoeningen.
- bilaterale oculaire betrokkenheid (A of b moet worden voldaan, afhankelijk van het stadium van de ziekte wanneer de patiënt wordt onderzocht).
- vroege manifestaties van de ziekte.
- Er moet bewijs zijn van een diffuse choroiditis (met of zonder voorafgaande uveïtis, vitreous inflammatory reaction, of optic disk hyperemia), die zich kan manifesteren als een van de volgende:
- focale gebieden van subretinale vloeistof, of
- bulleuze sereuze netvliesloslating.
- Met onduidelijke fundus bevindingen; beide van de volgende onderdelen moeten aanwezig zijn:
- concentratiegebieden is van een vertraging in de choroidale perfusie, multifocale gebieden van pinpoint lekkage, grote placoid gebieden van hyperfluorescence, pooling binnen subretinale vloeistof, en de oogzenuw vlekken (in volgorde van opeenvolgende verschijning) door fluoresceïne-angiografie en
- Diffuse choroidale verdikking, zonder bewijs van posterieure scleritis door echografie.
- Late manifestaties van de ziekte.
- voorgeschiedenis die wijst op eerdere aanwezigheid van bevindingen van 3a, en ofwel beide (2) en (3) hieronder of meerdere tekenen van (3):
- oculaire depigmentatie (een van de volgende manifestaties is voldoende): (a) Sunset glow fundus, of (B) Sugiura-teken.
- andere oculaire tekenen:
- nummulaire chorioretinale gedepigmenteerde littekens, of
- klonteren en/of migratie van het pigmentepitheel van de retina, of
- terugkerende of chronische voorafgaande uveïtis.
- neurologische / auditieve bevindingen (kunnen verdwenen zijn op het moment van onderzoek).
- Meningismus (malaise, koorts, hoofdpijn, misselijkheid, buikpijn, stijfheid van de nek en rug, of een combinatie van deze factoren; hoofdpijn alleen is echter niet voldoende om aan de definitie van meningismus te voldoen), of
- Tinnitus, of
- cerebrospinale vocht pleiocytose.
- integumentaire bevinding (niet voorafgaand aan het ontstaan van het centrale zenuwstelsel of oculaire ziekte).
- Alopecia, of
- poliose, of
- Vitiligo.
onvolledige ziekte van Vogt-Koyanagi-Harada (criteria 1 tot 3 en 4 of 5 moeten aanwezig zijn)
- geen voorgeschiedenis van penetrerend oculair trauma of chirurgie voorafgaand aan het begin van uveïtis, en
- geen klinische of laboratoriumgegevens die wijzen op andere entiteiten met een oculaire aandoening, en
- bilaterale oculaire betrokkenheid.
- neurologische / auditieve bevindingen; zoals gedefinieerd voor volledige ziekte van Vogt-Koyanagi-Harada hierboven, of
- integumentaire bevindingen; zoals gedefinieerd voor volledige ziekte van Vogt-Koyanagi-Harada hierboven.
waarschijnlijke ziekte van Vogt-Koyanagi-Harada (geïsoleerde oculaire ziekte; criteria 1 tot 3 moeten aanwezig zijn)
- geen voorgeschiedenis van penetrerend oculair trauma of chirurgie voorafgaand aan het begin van uveïtis.
- geen klinische of laboratoriumgegevens die wijzen op andere entiteiten met oculaire aandoeningen.
- bilaterale oculaire betrokkenheid zoals gedefinieerd voor volledige ziekte van Vogt-Koyanagi-Harada hierboven. “
testen/laboratoriumwerk
bij het eerste werk van VKH moet men overwegen de volgende tests te verkrijgen:
- optische coherentietomografie (OCT): In de acute uveitische fase, OCT zal waarschijnlijk vertonen significante choroïdale verdikking en sereuze netvliesloslating. De subretinale vochtophopingen kunnen septaties hebben waarvan wordt aangenomen dat ze fibrinemembranen en ontstekingsproducten zijn, waardoor een lobulaire structuur ontstaat die ook kan worden gezien bij fluoresceïne angiografie. In de herstelfase, kan OCT gebieden van netvlies dunner na opgelost ontsteking na corticosteroid behandeling detecteren .
- B-scan echografie: In de acute fase, echografie kan diffuse posterieure choroïdale verdikking, posterieure sclerale verdikking, netvliesloslating, en glasvocht troebelingen tonen . Ciliaire effusies kunnen worden waargenomen met echografie biomicroscopie . Deze test is ook nuttig voor het uitsluiten van posterieure scleritis.
- fluoresceïne angiografie (FA): Klassiek onthult FA multifocale choroïdale hypofluorescerende punten in de vroege fase, gevolgd door meerdere focale hyperfluorescerende gebieden met diffuse lekkage in de late fase . De kleurstof lekt door RPE en accumuleert in de subretinale ruimte die de hyperfluorescent punten omringt. FA kan Diagnostisch nuttig zijn wanneer de ziekte van VKH zonder extraoculaire symptomen presenteert. Optische schijf hyperfluorescentie en vensterdefecten veroorzaakt door atrofische chorioretinale littekens kunnen worden gezien in de mid-periferie . FA in de chronisch-terugkerende fase van de ziekte van VKH vertoont niet-specifieke vensterdefecten als gevolg van RPE-schade, choroïdale neovascularisatie en subretinale fibrose .
- angiografie van Indocyaninegroen (ICG) : Vroege fase ICG toont hyperfluorescente stromale vaten die wijzen op choroïdale vasculopathie en hypofluorescente donkere punten die overeenkomen met granulomen en vertraagde fragmentarische vulling van choroïdale vasculatuur . De late fase onthult vage stromale vasculaire patronen en diffuse choroïdale hyperfluorescentie. Disc hyperfluorescentie is suggestief van ernstige ziekte. ICGA kan subklinische choroïdale ontsteking in zeer vroege stadia of zelfs na systemische therapie detecteren .
- lumbale punctie: pleocytose in het cerebrospinale vocht is aanwezig bij de meerderheid van de VKH-patiënten. Lumbaalpunctie dient vroeg in het ziekteverloop te worden uitgevoerd, aangezien pleocytose kan verdwijnen
behandelings-/behandelrichtlijnen
behandelingsdoelen bij VKH omvatten vroege diagnose en onderdrukking van actieve ontsteking, samen met preventie van recidiverende ontsteking en visiebedreigende complicaties, zoals glaucoom, loslating van de bulleuze retina en choroïdale neovascularisatie.
systemische corticosteroïdbehandeling is de voorkeurstherapie voor VKH-ziekte, vooral tijdens het acute uveitische Stadium. Het is aangetoond dat de toedieningsroute van corticosteroïden (oraal versus intraveneus) geen invloed heeft op de gezichtsscherpte of het optreden van visueel significante complicaties bij de behandeling van acute VKH . Voor ernstige ziekten is het voorgestelde protocol intraveneuze toediening van methylprednisolon gedurende drie dagen, gevolgd door orale behandeling met hoge doses prednison. Bij milde tot matige ziekte kan een hoge dosis oraal prednison voldoende zijn bij 1-2 mg / kg / dag. De steroïde dosis moet langzaam worden afgebouwd gedurende ongeveer zes maanden om herhaling te voorkomen . De agressieve vroege behandeling, naast het periodieke fa testen die verdwijning van kleurstoflekkage door RPE tonen, kan helpen om verdere ziekteprogressie, herhaling, en extraocular manifestaties te verhinderen . Topische steroïden en cycloplegica kunnen cellen in de voorste oogkamer en glasvochthumor verminderen.
intravitreale en sub-Tenoninjecties van triamcinolon zijn gebruikt voor kortdurende controle van intraoculaire ontsteking tijdens de acute of terugkerende fasen; deze lokale therapieën moeten worden overwogen in het geval van recalcitrant ziekte en bij patiënten die slecht verdragen de ongunstige systemische bijwerkingen van steroïden gezien de uitgebreide steroïde taper. Intravitreale anti-VEGF injecties worden soms gebruikt voor controle van choroïdale neovascularisatie en in gevallen van aanhoudende foveale sereuze netvliesloslating .
Steroïdsparende middelen, waaronder antimetabolieten, calcineurineremmers, Biologica, TNF-alfa-remmers of cytotoxische middelen, kunnen worden gebruikt voor de behandeling van VKH en moeten zorgvuldig worden gecontroleerd, vaak in coördinatie met een reumatologische dienst . Er is lopende discussie geweest over het gebruik van niet-steroïde immunosuppressiva als eerstelijnstherapie voor de ziekte van VKH. Echter, een recente studie toonde geen verschillen in resultaten tussen vroege eerstelijns immunomodulerende behandeling (IMT) en prednison behandeling alleen . Verder zijn immunosuppressieve en biologische therapieën duur en vereisen ze een zorgvuldige evaluatie voorafgaand aan de behandeling en frequente follow-up met bloedonderzoek om ernstige bijwerkingen te beoordelen.
in het chronisch recidiverende stadium kan frequent recidief duiden op resistentie tegen corticosteroïdtherapie en op de noodzaak van steroïdsparende immunomodulerende behandeling . De aangewezen agent voor steroid-resistente herhaling of steroid-intolerantie is cyclosporine . Infliximab, rituximab, adalimumab en interferon alfa-2a zijn biologische middelen die ook zijn gebruikt voor de behandeling van refractaire uveïtis bij de ziekte van VKH.
voor de behandeling van uveïtis anterior, vaak geassocieerd met acute VKH, dienen topische steroïden (bijv. prednisolonacetaat 1%) en topische cyloplegie (bijv. cyclopentolaat 1% of atropine 1%) te worden voorgeschreven, afhankelijk van de mate van ontsteking van de voorste oogkamer.
oculaire complicaties worden vaak geassocieerd met de ziekte van VKH. Gezien de vele stadia en de verscheidenheid van presentaties waarin een patiënt met VKH kan presenteren, kan de behandeling in veel gevallen worden uitgesteld. Bij ernstige vormen van VKH en bij recidieven, kan intraoculaire ontsteking moeilijk te controleren zijn en kan leiden tot structurele schade. Meer dan 50% van de patiënten ontwikkelt gerelateerde complicaties, waaronder cataract, secundair glaucoom, choroïdale neovasculaire membranen, subretinale fibrose of een combinatie hiervan (Figuur 6) .
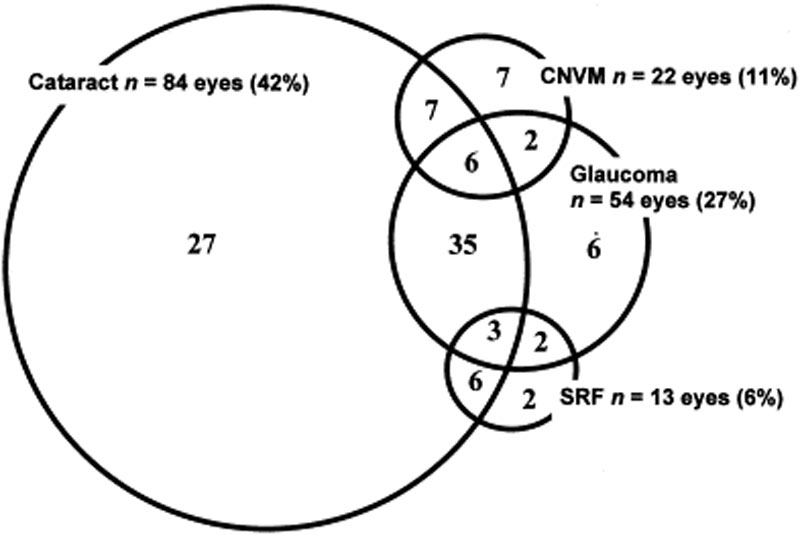
Figuur 6: Venn-diagram met complicaties bij VKH-patiënten. (Gebruikt met toestemming van Am J Ophthalmol. 2001;131(5):599-606 )
epidemiologie en etiologie
- typische aanvangsleeftijd: 20 tot 50 jaar
- vrouw > man
- predispositie in populaties met gepigmenteerde huid, met name Aziatisch, Midden-Oosten, Latijns-Amerikaans en inheemse Amerikanen
- genetische risico ‘ s: HLA-DR1, HLA-DR4, HLA-DRB1*0405 alleles
- Possible autoimmune targeting of melanocyte antigens
- Possible viral molecular mimicry as inciting factor in autoinflammatory activation
SIGNS
- Prodromal: Pleocytosis in CSF, meningitis, vertigo
- Uveitic: Bilateral granulomatous anterior or multifocal uveitis, multifocal serous retinal detachments in both eyes, optic nerve head edema and hyperemia
- Convalescent: Alopecia, poliosis, sunset glow fundus, Sugiura sign (limbale depigmentatie), Dalen-Fuchs knobbeltjes onder de RPE
- chronisch recidiverend: Herhaling van uveitis anterior, subretinale fibrosis, choroidale neovascular membranen, multifocale chorioretinal littekens, secundair glaucoom, staar
KLACHTEN
- Influenza-achtige prodromale met hoofdpijn, koorts, malaise, misselijkheid en/of braken
- gehoorverlies, tinnitus en dysacusis
- Nek stijfheid
- snelle verslechtering van wazig zien, waarbij beide ogen
- roodheid van het Oog, pijn, drijvers en/of fotofobie
BEHANDELING/MANAGEMENT
- Hoge-dosis systemic corticosteroids (intravenous vs oral) for the acute phase with slow taper
- Immunosuppressive agents (cyclosporine) and biologics (infliximab) for recurrent disease refractory to corticosteroids
- Topical steroids (prednisolone acetate 1%) and cycloplegics (cyclopentolate 1% or atropine 1%) for anterior uveitis and ciliary spasm
- Intravitreal triamcinolone and/or intravitreal anti-VEGF injections for control of inflammation, choroidal neovascular membrane, and/or persistent subfoveal fluid
- Du L, Kijlstra a, Yang P. Vogt-Koyanagi-Harada disease: Novel insights into pathophysiology, diagnosis and treatment. Prog Retin Eye Res 2016; 52: 84-111. https://PubMed.gov/26875727. DOI: 10.1016 / j. preteyeres.2016.02.002
- Yamaki K, Gocho K, Hayakawa K, Kondo i, Sakuragi S. tyrosinase familie eiwitten zijn antigenen specifiek voor de ziekte van Vogt-Koyanagi-Harada. J Immunol 2000; 165(12): 7323-7329. https://PubMed.gov/11120868
- Horie Y, Takemoto Y, Miyazaki A, Namba K, Kase S, Yoshida K, Ota m, Hasumi Y, Inoko H, Mizuki N, Ohno S. Tyrosinase genfamilie en de ziekte van Vogt-Koyanagi-Harada bij Japanse patiënten. Mol Vis 2006; 12: 1601-1605. https://PubMed.gov/17200659
- Ng JY, Luk FO, Lai TY, Pang CP. Invloed van Moleculaire Genetica bij de ziekte van Vogt-Koyanagi-Harada. J Oftalmische Inflamm Infecteren 2014; 4: 20. https://PubMed.gov/25097674. DOI: 10.1186 / s12348-014-0020-1
- Bowling B. uveïtis. Kanski ‘ s Clinical Ophthalmology New York, New York: Elsevier; 2016; chapter 11; p. 395-465.
- Yeh PT YC, Yang CH, Lin CP. Nonrhegmatogene Netvliesloslating. In: Schachat AP SS, Hinton DR, Wilkinson CP, Wiedemann P,, editor. Ryan ‘ s netvlies. New York: Elsevier; 2018; hoofdstuk 99; p. 1828-1849.
- Goto H RK, ziekte van Rao N. Vogt–Koyanagi–Harada. In: Schachat AP SS, Hinton DR, Wilkinson CP, Wiedemann P, editor. Ryan ‘ s netvlies. New York, New York: Elsevier; 2018; hoofdstuk 78; p. 1505-1515.Riddington L, Hall AJ, Tait B, Nicholson I, Varney M. Vogt-Koyanagi-Harada syndroom bij patiënten van Vietnamese afkomst. Aust N Z J Ophthalmol 1996;24 (2): 147-149. https://PubMed.gov/9199747
- Sugita s, Takase H, Kawaguchi T, Taguchi C, Mochizuki M. Kruisreactie tussen tyrosinasepeptiden en cytomegalovirusantigeen door T-cellen van patiënten met de ziekte van Vogt-Koyanagi-Harada. Int Ophthalmol 2007; 27 (2-3): 87-95. https://PubMed.gov/17253112. DOI: 10.1007/s10792-006-9020-y
- Freund BK SD, Mieler WF, Yannuzzi LA. Ontsteking. De Netvliesatlas. New York, New York: Elsevier 2017; hoofdstuk 4; p. 279-398.de ziekte van Rao N. Vogt-Koyanagi-Harada. In: J YMaD, redacteur. Oogheelkunde. New York, New York: Elsevier; 2014; hoofdstuk 7.17; p. 761-763.
- Rao NA, Xu S, lettertype RL. Sympathische oftalmie. Een immunohistochemische studie van epithelioïde en reuzencellen. Oftalmologie 1985; 92 (12):1660-1662. https://PubMed.gov/4088616
- Nussenblatt RB. Syndroom Van Vogt-Koyanagi-Harada. In: Whitcup RBNaSM, editor. Uveïtis: grondbeginselen en klinische praktijk. 4e editie ed: Elsevier; 2010; hoofdstuk Hoofdstuk 24.Read RW, Holland GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L, Pivetti-Pezzi P, Tessler HH, Usui M. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature. Am J Ophthalmol 2001; 131 (5): 647-652. https://PubMed.gov/11336942
- Chung h, Choi DG. Klinische analyse van uveïtis. Koreaans J Ophthalmol 1989; 3 (1):33-37. https://PubMed.gov/2795939. DOI: 10.3341 / kjo.1989.3.1.33
- Abu El-Asrar AM, Al-Kharashi AS, Aldibhi H, Al-Fraykh H, kangave D. Vogt-Koyanagi-Harada disease in children. Oog (Lond)) 2008;22(9):1124-1131. https://PubMed.gov/17479116. DOI: 10.1038 / sj.oog.6702859
- Martin TD, Rathinam SR, Cunningham ET. Prevalentie, klinische kenmerken en oorzaken van verlies van het gezichtsvermogen bij kinderen met de ziekte van Vogt-Koyanagi-Harada in Zuid-India. Retina 2010; 30 (7): 1113-1121. https://PubMed.gov/20168275. DOI: 10.1097 / IAE.0b013e3181c96a87
- Forster DJ, Green RL, Rao NA. Unilaterale manifestatie van het syndroom van Vogt-Koyanagi-Harada bij een 7-jarig kind. Am J Ophthalmol 1991; 111 (3):380-382. https://PubMed.gov/2000916
- Yamamoto Y, Fukushima a, Nishino K, Koura Y, Komatsu T, Ueno H. ziekte van Vogt-koyanagi-harada met aanvang bij oudere patiënten in de leeftijd van 68 tot 89 jaar. Jpn J Ophthalmol 2007; 51 (1): 60-63. https://PubMed.gov/17295144. DOI: 10.1007/s10384-006-0379-0
- Wang Y, Chan CC. Geslachtsverschillen bij de ziekte van vogt-koyanagi-harada en sympathische oftalmie. J Ophthalmol 2014; 2014: 157803. https://PubMed.gov/24734166. DOI: 10.1155/2014/157803
- Nakao K, Abematsu N, Mizushima Y, Sakamoto T. zwelling van de optische schijf bij de ziekte van Vogt-Koyanagi-Harada. Invest Ophthalmol Vis Sci 2012; 53 (4): 1917-1922. https://PubMed.gov/22408010. DOI: 10.1167 / iovs.11-8984
- Rao NA, Gupta A, Dustin L, Chee SP, Okada AA, Khairallah M, Bodaghi B, Lehoang P, Accorinti M, Mochizuki m, Prabriputaloong T, Read RW. Frequentie van onderscheidende klinische kenmerken bij de ziekte van Vogt-Koyanagi-Harada. Ophthalmology 2010;117(3):591-599, 599.e591. https://PubMed.gov/20036008. DOI: 10.1016/j.ophtha.2009.08.030
- Veerappan M, Fleischman D, Ulrich JN, Stinnett SS, Jaffe GJ, Allingham RR. The Relationship of Vogt-Koyanagi-Harada Syndrome to Ocular Hypertension and Glaucoma. Ocul Immunol Inflamm 2017;25(6):748-752. https://PubMed.gov/27438521. DOI: 10.1080/09273948.2016.1189578
- Baltmr A, Lightman S, Tomkins-Netzer O. Vogt-Koyanagi-Harada syndrome – current perspectives. Clin Ophthalmol 2016;10:2345-2361. https://PubMed.gov/27932857. DOI: 10.2147/OPTH.S94866
- Kitaichi n, Matoba H, Ohno S. the positive role of lumbar punction in the diagnosis of Vogt-Koyanagi-Harada disease: lymphocyt subsets in the watery humor and cerebrospinal fluid. Int Ophthalmol 2007; 27 (2-3):97-103. https://PubMed.gov/17211585. DOI: 10.1007/s10792-006-9016-7
- Oshima Y, Harino S, Hara Y, Tano Y. indocyanine green angiographic findings in Vogt-Koyanagi-Harada disease. Am J Ophthalmol 1996; 122(1): 58-66. https://PubMed.gov/8659599
- Read RW, Yu F, Accorinti M, Bodaghi B, Chee SP, Fardeau C, Goto H, Holland GN, Kawashima H, Kojima E, Lehoang P, Lemaitre C, Okada AA, Pivetti-Pezzi P, Secchi A, See RF, Tabbara KF, Usui M, Rao NA. Evaluatie van het effect op de resultaten van de toedieningsroute van corticosteroïden bij de acute ziekte van Vogt-Koyanagi-Harada. Am J Ophthalmol 2006;142 (1): 119-124. https://PubMed.gov/16815259. DOI: 10.1016 / j. ajo.2006.02.049
- Rubsamen PE, Gass JD. Syndroom van Vogt-Koyanagi-Harada. Klinische cursus, therapie, en langdurige visuele uitkomst. Arch Ophthalmol 1991;109(5):682-687. https://PubMed.gov/2025171
- Urzua CA, Velasquez V, Sabat P, Berger O, Ramirez S, Goecke A, Vásquez DH, Gatica H, Guerrero J. eerdere immunomodulerende behandeling wordt geassocieerd met betere visuele resultaten in een subgroep van patiënten met de ziekte van Vogt-Koyanagi-Harada. Acta Ophthalmol 2015; 93 (6): e475-480. https://PubMed.gov/25565265. DOI: 10.1111 / aos.12648
- Read RW, Rechodouni A, Butani N, Johnston R, LaBree LD, Smith RE, Rao NA. Complicaties en prognostische factoren bij de ziekte van Vogt-Koyanagi-Harada. Am J Ophthalmol 2001; 131 (5): 599-606. https://PubMed.gov/11336934
Suggested Citation Format
Mai AP, Tran C, Wilson CW, Fox AR, Boldt HC. De ziekte van Vogt-Koyanagi-Harada (VKH). EyeRounds.org. April 1, 2019. Beschikbaar vanaf http://EyeRounds.org/cases/284-vogt-koyanagi-harada.htm
laatst bijgewerkt op: 01-04-2019 - vroege manifestaties van de ziekte.
Leave a Reply