Vogt-Koyanagi-Haradův (VKH) Onemocnění
Autoři: Anthony P. Mai, BS; Charlene Tran, BS; Caroline W. Wilson, MD; Austin, R. Fox, MD; H. Culver Boldt, MD,
1. dubna, 2019
POČÁTEČNÍ PREZENTACE
Hlavní Stížnost
Rozmazané vidění a bolesti hlavy
Historie Současné Nemoci
44-rok-staré Vietnamské ženy prezentovány na pohotovost s 10-denní historie o progresivní rozmazané vidění v obou očích, a tři-denní historie o silné bolesti hlavy. Její centrální ztráta zraku se nezlepšila lom jejího optometristy. Její těžké týlní bolesti hlavy se zhoršovaly pohybem a byly spojeny s generalizovanou malátností, extrémní únavou, mírnou fotofobií a slzami. Acetaminofen částečně zmírnil bolest.
nedávno odcestovala do Vietnamu, ale popřela, že by se tam setkala s nemocnými kontakty. Popřela klaudikaci čelistí, horečky nebo změny hmotnosti. Popřela kožní vyrážky, změny sluchu, tinnitus, závratě, necitlivost nebo brnění. Popřela, že by někdy měla tuberkulózu. Neměla žádné předchozí problémy se zrakem, autoimunitní stavy nebo rakovinu.
Kolem Oční Historie
- Historie kosmetické operace očních víček (bilaterální blefaroplastika) tři roky před
- Žádná historie oční trauma nebo onemocnění
Zdravotní Minulost
None
Léky
Acetaminophen, jak je potřeba,
Alergie
Žádné známé lékové alergie
Rodinné Historie
Žádná historie oční onemocnění nebo autoimunitní onemocnění,
Sociální Historie
Ona se přistěhoval z Vietnamu několik let před prezentaci. Je vdaná a má tři děti. Pracuje v nehtovém salonu. Nekonzumuje tabákové výrobky, alkohol ani nedovolené látky. Do Vietnamu cestuje každých šest až dvanáct měsíců.
přehled Systémů
Negativní, až na to, co je uvedeno v historie současné nemoci
OČNÍ VYŠETŘENÍ
Zraková Ostrost s/bez korekce (Snellen)
- Pravé oko (OD): 20/300 (žádné zlepšení s pinhole)
- Levé oko (OS): 20/60-2+2 (žádné zlepšení s pinhole)
Pohyblivosti Oka/Zarovnání
Plné okohybné pohyby v obou očích (OU)
Nitroočního Tlaku (IOP): (Tonopen)
- OD: 12 mmHg
- OS: 14 mmHg
Žáci
- OD: 4 mm v tmavě, 3 mm ve světle, ne relativní aferentní pupilární defekt (RAPD)
- OS: 4 mm v tmavě, 3 mm v světle, ne RAPD
Konfrontace vizuální pole: (Počet prstů)
- OD: Centrální skotom
- OS: Celkové inferotemporálním vada
Externí
Normální na obou stranách
Štěrbinové lampy vyšetření
- Víčka/řasy: Normální OU
- Spojivky/skléry: Jasné a klidné OU
- Rohovky: 1+ tečkovité epiteliální eroze, ne keratic sráží OU
- Přední komory: Trace mobilní a světlice a hluboké OU
- Iris: Normální architektura OU
- Objektiv: Čirý OU
Rozšířené vyšetření očního pozadí (DFE)
- Sklivce: Stopy předního sklivce buňkami OU
- Disk:
- OD: Stupeň 3 disk edém, hyperemická
- OS: Stupeň 2-3 disk edém, hyperemická
- Cup-to-disc ratio: 0.0 OU
- Macula:
- OD: 3+ cystoidní makulární edém (CME) a subretinálních tekutiny (SRF) rozšíření z disku do spánkového makuly. Žádné lipidy ani exsudáty. Bažinatý choroid.
- OS: 2+ CME a SRF probíhající z disku přes fovea. 1-2 + lineární lipid probíhající od disku směrem k fovea. Bažinatý choroid.
- Lodě:
- OD: Opláštění časně
- OS: Normální
- Periferie:
- OD: Cystická sítnice chomáč přední s rovníkem v 10:30
- OS: Mělké SRF přední rovníku na 4:00
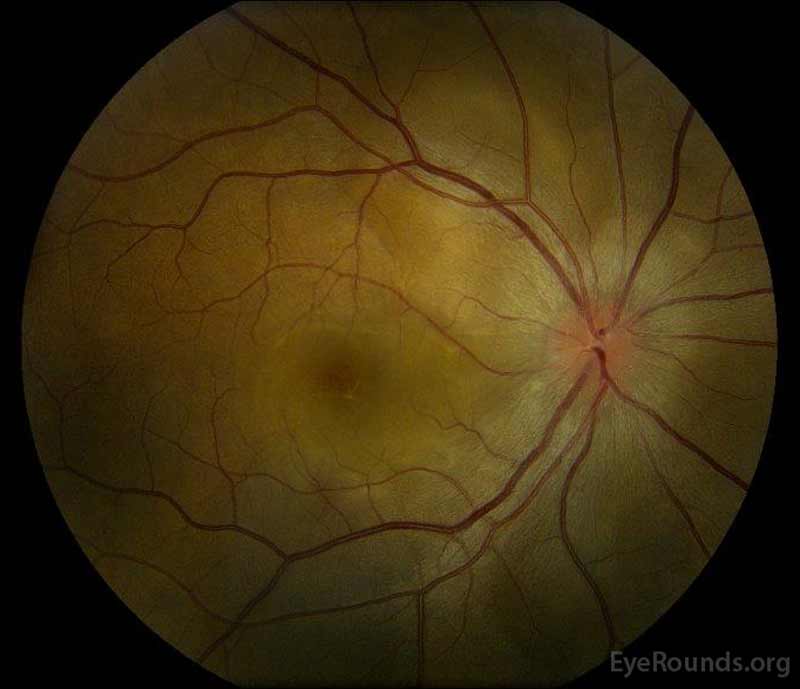 |
 |
Obrázek 1: Barevné fundus fotografií v prezentaci: (Levý obrázek), pravé oko má disk edém a mírné překrvení, stejně jako subretinálních kapaliny vystupující z disku časně přes macula. K dispozici je také fokální serózní oddělení sítnice superotemporální k disku, podél nadřazené arkády. (Right image) The left eye has disc edema and mild hyperemia, along with subretinal fluid extending from the disc to the macula and linear lipid deposits in the nasal macula.
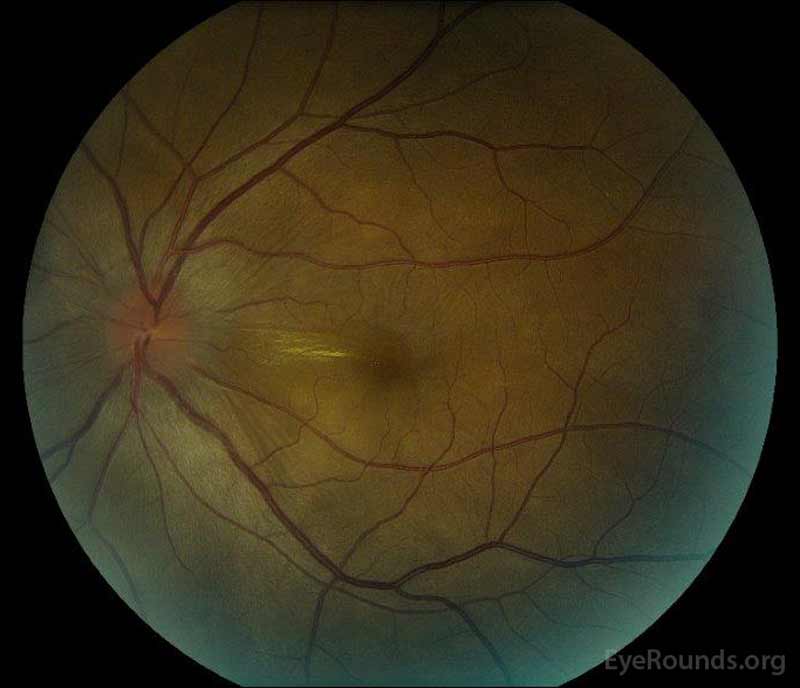
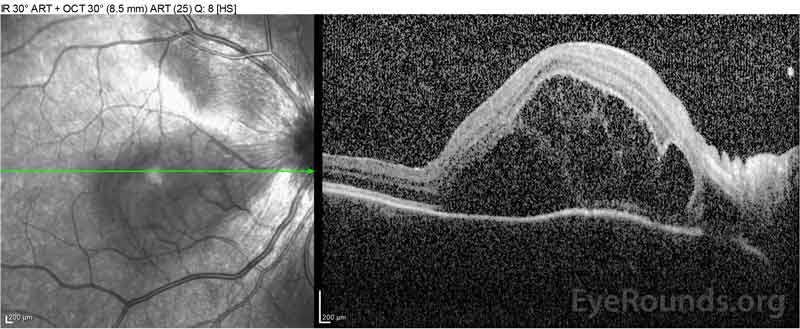 |
 |
 |
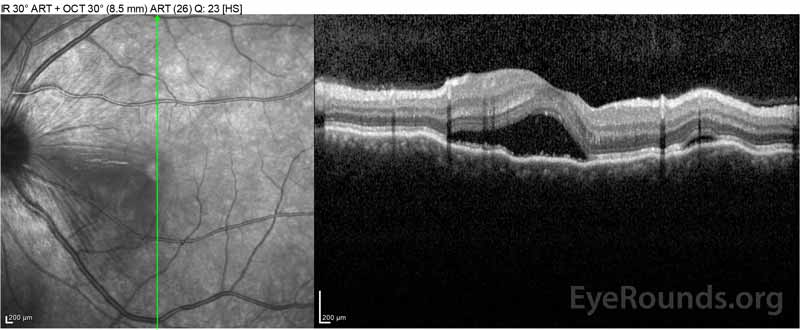 |
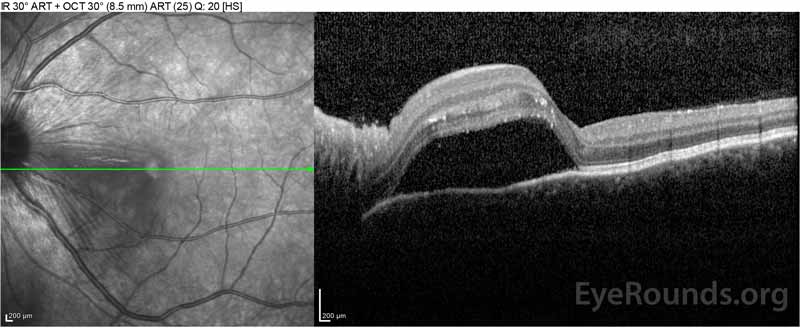
Figure 2: Optická koherenční tomografie (OCT) pravého oka (horní panely) ukazuje serózní odchlípení sítnice zahrnující fovea s rozsáhlými nadložních intraretinal tekutin, narušení zevní retinální vrstvy, a vlnění zahuštěný cévnatky. Říjen levého oka (spodní panely) ukazuje serózní oddělení sítnice v nosní makule sahající až k fovea.
 |
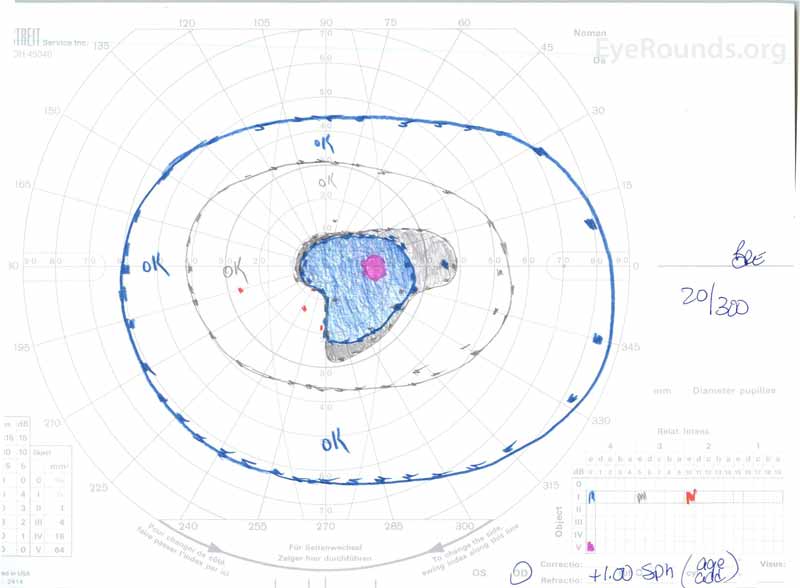 |
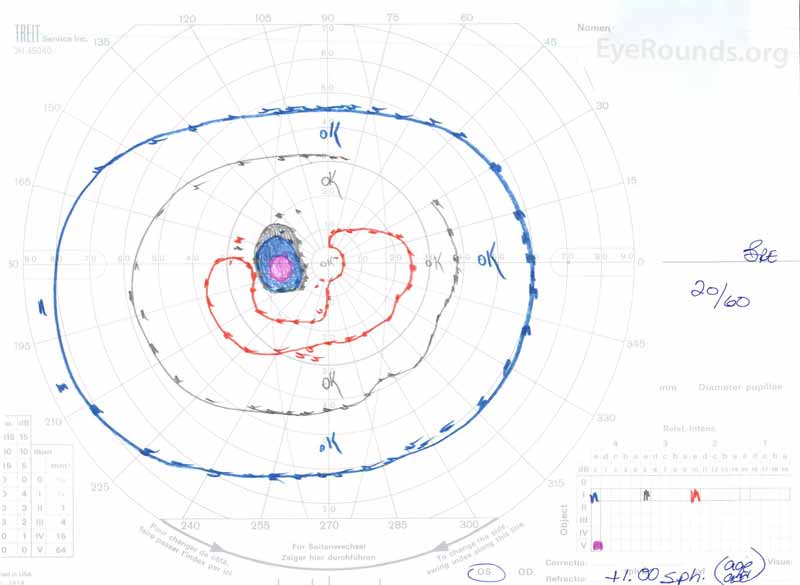
Obrázek 3: Goldman vizuální pole (GVF), OU. (Levý obrázek) OS ukazuje zvětšené fyziologické slepé místo a mírný centrální skotom. (Obrázek vpravo) od ukazuje středně hustý centrální skotom zahrnující fyziologické slepé místo a rozšiřující se inferotemporálně.
B-scan: Žádné známky skleritida, mírné vitreal zákaly/buňky ještě zajisti
Diferenciální Diagnóza
- Akutní zadní multifokální placoid pigment epitheliopathy (APMPPE)
- Centrální serózní chorioretinopathy
- zánět Zrakového nervu
- Panuveitis
- Autoimunitní onemocnění (např. LUPUS, sarkoidóza)
- Infekce (e.g., syfilis, tuberkulóza, Bartonella henselae)
- Malignity (např. nitrooční lymfom)
- Zadní skleritida
- Sympatické oftalmie
- Uveal výpotek syndrom
- Vogt-Koyanagi-Harada Syndrom
PRÁCE
Kompletní krevní obraz
počet Bílých krvinek: 4.9 K/mm3 (Ref: 3.7-10.5)
počet Červených krvinek 3.99 M/mm3 (Ref: 4.0-5.2)
Hemoglobin 11,6 g/dL (Ref: 11.9-15.5)
Hematokritu 35 % (Ref: 35-47)
Basic metabolic panel
Sodium 138 mEq/L (Ref: 135-145)
Potassium 4.3 mEq/L (Ref: 3.5-5.0)
Chloride 107 mEq/L (Ref: 95-107)
CO2 20 mEq/L (Ref: 22-29)
Blood urea nitrogen 16 mEq/dL (Ref: 10-20)
Creatinine 0.7 mg/dL (Ref: 0.5-1.0)
C-reactive Protein (CRP): <0.5 mg/dL (Ref: <=0.5)
Erythrocyte sedimentation rate (ESR): 12 mm/Hr (Ref: 0-20)
Angiotensin–converting enzyme (ACE): 13 U/L (Ref: 8-52)
příbalový leták QuantiFERON-TB Gold: negativní
Železo, krevní 54 mikrogramů/dL (Ref: 37-145)
Celkového železa vazebné kapacity 379 mikrogramů/dL (Ref: 250-425)
KLINICKÉ KURZ
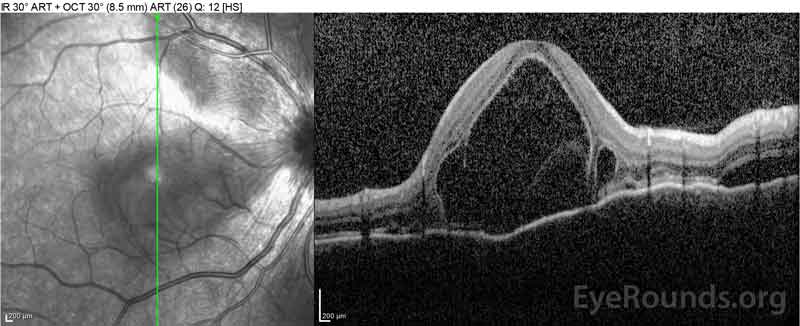
pacient byl zpočátku hodnocena pohotovost vzhledem k její stížnosti nový nástup silné bolesti hlavy a ztrátě zraku. Počítačová tomografie mozku (CT) a zobrazování magnetickou rezonancí (MRI) byly všední. ESR a CRP byly v normálních hladinách. Oční klinika ji následující den vyhodnotila a zjistila bilaterální serózní oddělení sítnice a panuveitidu. ACE a QuantiFERON-TB Gold labs byly oba negativní. Byla diagnostikována Vogt-Koyanagi-Harada nemoc na základě jejího klinického projevu a asijského původu. Byla léčena 80 mg prednisonu denně, acetaminofen podle potřeby pro bolesti hlavy a doplnění vitaminu D a vápníku. Její bolesti hlavy rychle ustoupily, a její zraková ostrost se během následujících dvou týdnů neustále zlepšovala. Její dávka prednisonu se poté během tří týdnů snížila na 40 mg s pokračujícím ústupem příznaků a zlepšením zrakové ostrosti. Během zúžení prednisonu se u ní neobjevily bolesti hlavy ani zhoršení zraku. Při jejím posledním jmenování, každý druhý den se zúžila na 5 mg, bez návratu příznaků. Její zrakové ostrosti na to, že následná návštěva byla 20/15-2 OD a 20/20+2 OS, a makulární OCT ukázal v plném rozlišení na disku edém a serózní odchlípení oddíly na obou očích (Obrázek 4).
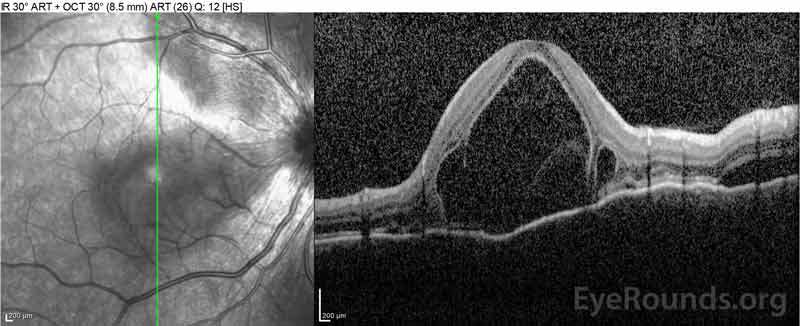
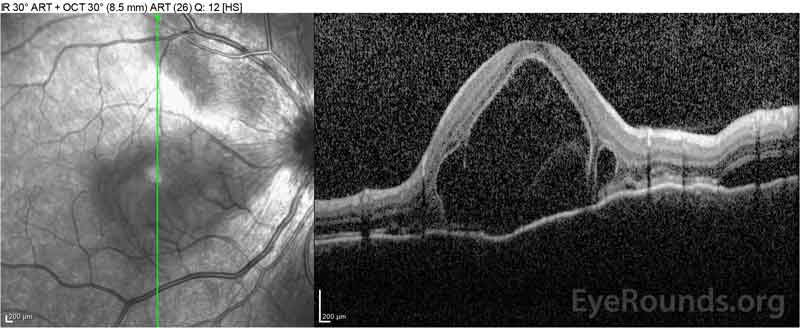
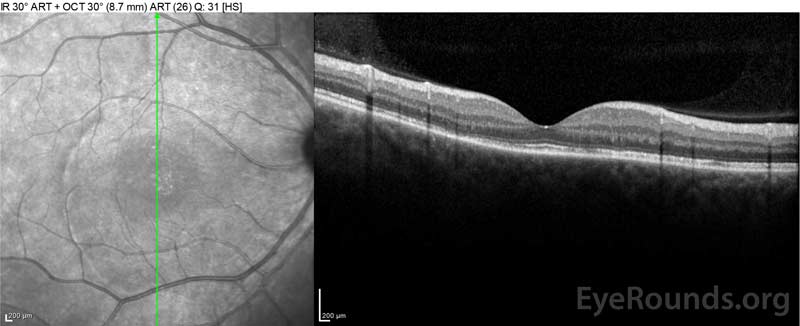
Obrázek 4: Optická koherentní tomografie ukazující subretinální tekutinu na začátku (nahoře) a průběh rozlišení v jednom týdnu (uprostřed) a pět týdnů (dole) při vysoké dávce perorálního zúžení prednisonu. Všimněte si vyhlazení choroidálních zvlnění léčbou.
 |
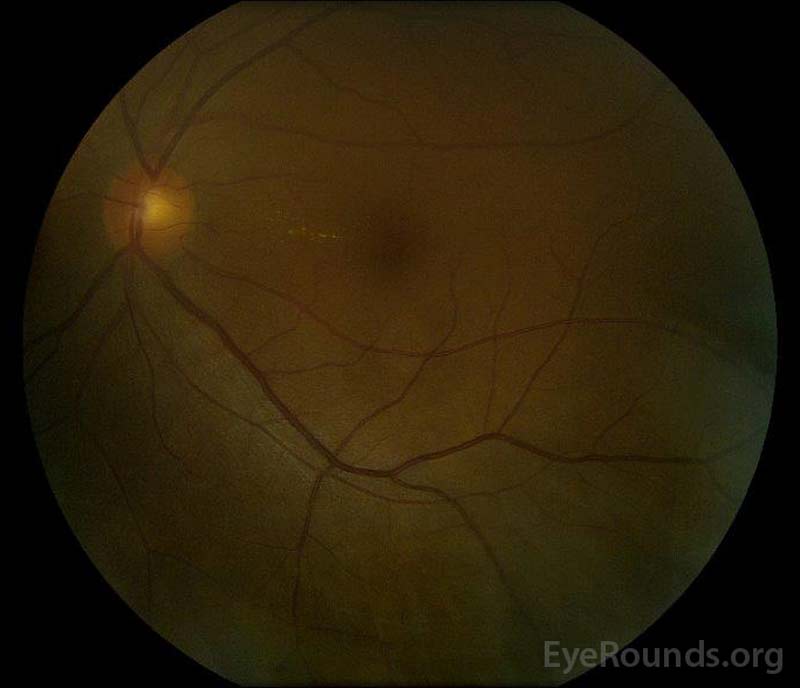 |
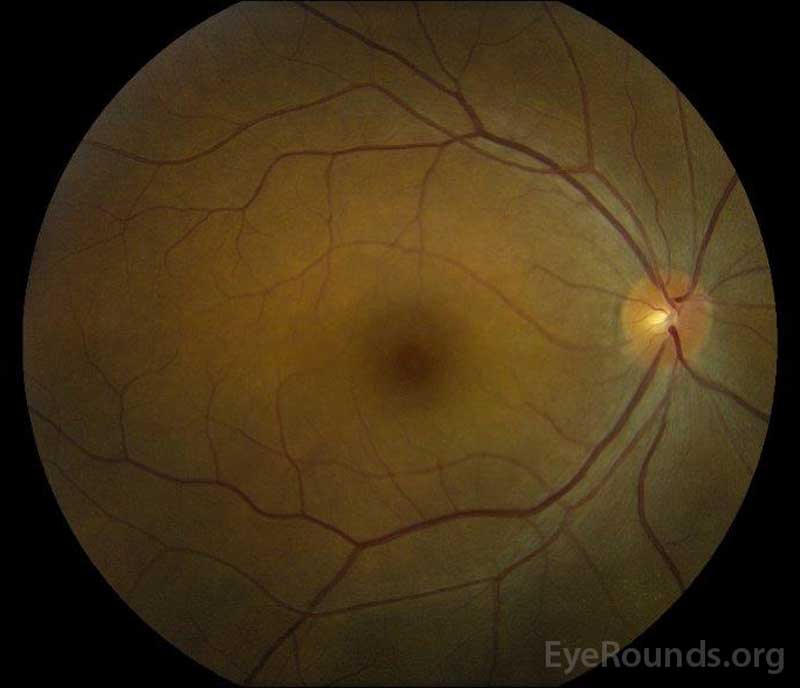
Obrázek 5: Barevné fotografie fundu práva (a) a levém (B) oči během rekonvalescence fáze prokazování zlepšení v ny tekutiny a disk edém.
DIAGNOSTIKA
Neúplné Vogt-Koyanagi-Haradova Choroba
DISKUSE
Vogt-Koyanagi-Haradův (VKH) Choroba je systémové autoimunitní onemocnění charakterizované bilaterální non-nekrotizující granulomatózní panuveitis spojené s okohybné kožní změny, jako obrnózou a vitiligo, a zánět postihující uvea, vnitřní ucho, vlasy, a plen. Harada nemoc je izolovaná uveitida bez systémových příznaků nebo příznaků VKH.
etiologie
etiologie onemocnění VKH je i přes současné výzkumné úsilí stále do značné míry neznámá. Předpokládá se, že se jedná o získané autoimunitní onemocnění zahrnující přecitlivělost zprostředkovanou T-buňkami na melanocytární vlastní antigeny, se základní genetickou predispozicí a možným mikrobiálním spouštěčem . Tyrosináza a peptidy související s tyrosinázou jsou antigeny melanocytů, které byly navrženy jako cíle autoimunitních procesů ve VKH . Podle jedné studie však zvýšené riziko onemocnění VKH nebylo spojeno s rodinou genů tyrosinázy .
vzhledem ke zvýšené prevalenci u určitých etnických a genderových skupin se předpokládá genetická predispozice v patogenezi VKH. Více genů, včetně genů lidského leukocytového antigenu (HLA) a interleukinu (IL), bylo spojeno s VKH v různých etnických populacích . Hla receptory jsou hlavní histokompatibilní komplexy u lidí, které představují peptidy imunitnímu systému. Hla-DR1, HLA-DR4, HLA-DRB1*0405 a HLA-DRw53 je několik haplotypů nalezených u pacientů s onemocněním VKH . HLA-DR4 je častější u japonských a hispánských lidí, zatímco HLA-DRB1 * 0405 je častější u korejských a blízkovýchodních pacientů . U vietnamských pacientů se vyskytují alely HLA-DR4 i HLA-DRB1*0405 . Receptor HLA-DRB1 se váže na antigeny melanocytů v různých kapacitách. Navzdory těmto sdružením se genetické testování v tuto chvíli nedoporučuje.
Vzhledem k obvyklé prodromální příznaky, které doprovázejí VKH, včetně horečky, bolesti hlavy, meningismus a hučení v uších, podněcování virové etiologie bylo navrhl jako podnět pro VKH nástup přes mechanismus molekulární mimikry u geneticky predisponovaných pacientů. Cytomegalovirový obal glykoprotein H má významnou aminokyselinovou homologii k tyrosinázovému peptidu a infekce CMV může vyvolat VKH prostřednictvím molekulární mimikry (tj. Byl také zapojen virus Ebstein-bar (EBV). Neexistují však žádné definitivní důkazy týkající se virové etiologie VKH a zůstává nejasné, co spouští autoimunitní odpověď VKH .
patofyziologie
existují čtyři klasické fáze VKH, které mohou mít variabilní prezentace: prodromální, akutní uveitické, rekonvalescentní a chronické recidivující. Histopatologické změny obvykle začínají v akutní fázi .
akutní uveitická fáze je charakterizována bilaterálním uveálním zahušťováním sekundárním po granulomatózním zánětu. Granulomy se skládají z lymfocytů, makrofágů a epitelioidních a obřích buněk naplněných granulemi . Ačkoli epiteloidní buňky byly dříve považovány za změněné melanocyty, následná imunohistochemická studie místo toho naznačila původ z tkáňových makrofágů . Granulomy naplněn epiteloidní histiocyty, nazývané Dalen-Fuchs uzliny, může být často viděn mezi pigmentového epitelu sítnice (RPE) a Bruch membránou. Na uveal granulomatózní zánět vede k choroidální zahušťování a exsudativní odchlípení oddíly naplněné s bílkovinnou tekutiny. Kromě toho pleocytóza (i.e., zvýšený počet buněk) může být přítomen v přední komoře a sklivci .
rekonvalescentní fáze je označena depigmentace cévnatky a okohybné oblastech, včetně periokulární pokožka a vlasy. A depigmentovaná cévnatky proti světle zrakového nervu dává dojem „západ slunce záře“ fundus, což je klasickým rysem této fáze VKH . Kromě toho se Dalen-Fuchsovy uzliny stávají výraznějšími pod RPE v rekonvalescentní fázi .
chronické-recidivující fáze se vyznačuje sníženou choroidální tloušťka, rozlišení serózní odchlípení oddíly, chronické mírné vitritis, a opakující se granulomatózní zánět předního segmentu. Během této fáze se může vyvinout choroidální neovaskularizace (CNV) a subretinální fibróza a jsou indikátory závažné progrese onemocnění . Katarakta a sekundární glaukom jsou dalšími komplikacemi dlouhodobého nebo opakujícího se zánětu v této fázi .
epidemiologie
VKH je převládající u Ras s tmavším pigmentem kůže, zejména Asiatů, Jihoameričanů, Středního východu a domorodých Američanů. Onemocnění VKH představuje >10% uveitidy v těchto populacích . Pouze 1-4% případů uveitidy je považováno za sekundární k onemocnění VKH ve Spojených státech (7). Ve Spojených Státech, většina případů VKH bylo zjištěno, že ovlivňují jednotlivce, Asijské, Hispánské a/nebo indiánské slušné . Zajímavé je, že onemocnění VKH zřídka postihuje Afričany navzdory jejich tmavé pigmentaci . Výskyt onemocnění VKH se mezi rasovými podskupinami v sousedních zemích velmi liší . Například výskyt VKH v Koreji je pouze 2%, mnohem nižší než v Japonsku a Číně .
VKH má typický nástup od 20 do 50 let věku ; studie však naznačují, že 3.1-13,4% VKH jsou případy dětských pacientů a 10% případů jsou ≥65 let . Klasicky, předpokládá se, že VKH má zálibu v ženském pohlaví, a zatímco většina studií ukazuje, že VKH neúměrně postihuje ženy, několik studií prokázalo mužskou predispozici nebo žádnou genderovou predispozici .
známky / příznaky
jak bylo uvedeno výše, čtyři stadia onemocnění VKH jsou prodromální, uveitické, rekonvalescentní a chronické recidivující. Každá fáze vykazuje odlišné klinické rysy.
- prodromální: toto počáteční stadium se může projevit jako onemocnění podobné chřipce s převážně ústavními příznaky, jako je bolest hlavy, závratě, horečka, únava a/nebo nauzea. Byly hlášeny neurologické příznaky meningitidy, obrny kraniálních nervů a optické neuritidy, jakož i sluchové příznaky tinnitu, dysakuze a závratě . Fotofobie, rozmazané vidění, plováky a/nebo bolest očí obvykle začínají do 48 hodin od prodromálních příznaků . Prodromální fáze obvykle trvá několik dní až týdnů.
- akutní Uveitika: tato fáze zahrnuje rozmazané vidění, fotofobii, injekci spojivky a bolest očí. Může se jednat o mírnou přední uveitidu, která se zpočátku jeví jako negranulomatózní. Jednostranný nástup obvykle přechází na bilaterální zapojení během 1-2 týdnů. Může se vyvinout granulomatózní přední uveitida s keratickými sraženinami skopového tuku. Zadní zkoušku nálezy mohou zahrnovat zrakového nervu edém a hyperémie , multifokální oblasti choroiditis, více oblastí serózní odchlípení oddíly lokalizována v zadní fundus, choroidální zahušťování, vyzařující chorioretinální záhyby, a vitritis . Serózní odchlípení oddíly mohou tvořit cloverleaf vzor v zadní fundus a mohl postupovat k rozsáhlé bulózní oddíly v závažných případech . Akutní zánětlivý glaukom byl spojen s touto fází onemocnění a může se projevit mělkou přední komorou sekundární k edému ciliárního těla, napodobující uzavření s akutním úhlem . Doba trvání akutní uveitické fáze závisí na rychlé diagnostice a léčbě.
- chronická Uveitická nebo rekonvalescentní: tato fáze se obvykle vyvíjí několik týdnů po akutní fázi a je charakterizována vitiligo (např. Depigmentace v blízkosti rohovky limbus, známý jako Sugiura znamení, může být viděn jeden měsíc po nástupu nemoci ; nicméně, toto znamení je zřídka vidět mimo japonskou populaci . Choroidální depigmentace se obvykle vyskytuje během několika měsíců a má za následek jasně oranžovo-červenou barvu choroidu a klasický „sunset glow fundus“.“Sunset glow fundus je považován za nejdůležitější a prediktivní v diagnostice chronické VKH . V polovině periferie se mohou tvořit dobře definované, kulaté, nummulární chorioretinální jizvy. Chronická uveitická fáze obvykle trvá několik měsíců.
- chronická recidivující: Tato fáze je charakterizována opakujícími se epizodami granulomatózní přední uveitida s skopové tuku keratic sraženin, iris uzliny, depigmentace duhovky, zadní synechiae, zadní subkapsulární katarakta, sekundární glaukom, choroidální neovaskulární membrány, a, nakonec, ny fibróza a nummular chorioretinální atrofie . Chronická fáze se obvykle vyvíjí nejméně šest měsíců po počáteční prezentaci. Serózní oddělení sítnice přítomné během akutní a rekonvalescentní fáze se obvykle neopakují při agresivní léčbě kortikosteroidy .
Diagnostická Kritéria
nejnovější diagnostická kritéria, s názvem Revidovaná Diagnostická Kritéria (RDC) pro VKH, byly definovány v roce 1999 na První Mezinárodní Workshop na VKH . Ty jsou uvedeny v tabulce 1. RDC jsou užitečné v tom, že rozdělují VKH do tří různých diagnostických kategorií na základě fáze onemocnění, během které pacient představuje: úplné, neúplné a pravděpodobné. Tato kategorizace onemocnění umožňuje vhodnou a včasnou léčbu „pravděpodobného“ onemocnění, které může pomoci zabránit progresi k“ úplnému “ onemocnění.
zpracování pro jiné příčiny očního zánětu, infekční i auto-zánětlivé, jsou nezbytné. Ty mohou zahrnovat sedimentace erytrocytů (ESR), C-reaktivní protein (CRP), quantiferon-Zlato testování na tuberkulózu, rapid plasma reagin (RPR) na syfilis, inhibitory angiotenzin-konvertujícího enzymu (ACE) a hrudníku x-ray pro sarkoidóza, antinukleární protilátky (ANA), a p-/c-ANCA. Také, historii posledních otevřených očních poranění nebo nitrooční operaci musí být zaznamenány a pravděpodobně naznačuje, sympatické oftalmie (TAK) jako pravděpodobnější diagnóza vzhledem k velmi podobné prezentace a patofyziologie sdílené mezi TAK a VKH .
podporovat diagnózu VKH v nejasných případech, lumbální punkce může být provedena hledat lymfocytární a monocytární pleocytózou; nicméně, toto je zřídka činné klinicky. Osmdesát procent pacientů má pleocytózou v mozkomíšním moku (CSF) v rámci jednoho týdne a 97% pleocytózou do tří týdnů. Zvýšené hladiny imunitních buněk mohou trvat až osm týdnů po nástupu onemocnění . Profily povrchových markerů T-buněk jsou podobné mezi CSF a komorovým humorem, ale liší se od krve. To naznačuje schopnost CSF přesně odrážet uveální zánět u onemocnění VKH .
Tabulka 1. Revidovaná Diagnostická Kritéria pro Vogt-Koyanagi-Haradova Choroba
*Z Tabulky 1 v (15).
„Kompletní Vogtův-Koyanagiho-Haradova choroba (kritéria 1 až 5 musí být přítomen)
- Žádná historie pronikající oční trauma nebo chirurgický zákrok předchází počáteční nástup uveitidy.
- žádné klinické nebo laboratorní důkazy svědčící o jiných entitách očního onemocnění.
- bilaterální oční postižení (a nebo b musí být splněno v závislosti na stadiu onemocnění při vyšetření pacienta).
- časné projevy nemoci.
- musí být prokázána difuzní choroiditis (s nebo bez přední uveitida, skelná zánětlivé reakce, nebo optického disku překrvení), která se může projevit jako jednu z následujících možností:
- časné projevy nemoci.
- Kontaktní plochy z ny tekutiny, nebo
- Bulózní serózní odchlípení oddíly.
- S nejednoznačnými fundus zjištění; obě tyto musí být přítomny i:
- Kontaktní plochy, zpoždění v choroidální perfuze, multifokální oblasti určit úniku, velké placoid oblastí hyperfluorescence, sdružování do ny tekutiny, a zrakového nervu barvení (uvedeny v pořadí sekvenční vzhled) pomocí fluoresceinové angiografie, a
- Difuzní choroidální zahušťování, bez důkazů, zadní skleritida ultrasonograficky.
- pozdní projevy nemoci.
- Historie připomínající předchozí přítomnost nálezů z 3a, a to buď oba (2) a (3) níže, nebo více příznaky, z (3):
- Oční depigmentace (buď z těchto projevů je dostačující): (a) západ Slunce záře fundus, nebo (b) Sugiura podepsat.
- Další oční příznaky:
- Nummular chorioretinální depigmentované jizvy, nebo
- pigmentového epitelu Sítnice shlukování a/nebo migrace, nebo
- Recidivující nebo chronické přední uveitidy.
- neurologické / sluchové nálezy (mohly být vyřešeny časem vyšetření).
- Meningismus (malátnost, horečka, bolest hlavy, nevolnost, břišní bolesti, ztuhlost šíje a zad, nebo kombinace těchto faktorů; bolest hlavy sama o sobě není dostatečné pro splnění definice meningismus, nicméně), nebo
- Hučení v uších, nebo
- pleocytózou Mozkomíšního moku.
- nález kožního krytu (nepředchází nástupu centrálního nervového systému nebo očního onemocnění).
- alopecie nebo
- polióza nebo
- Vitiligo.
Neúplné Vogt-Koyanagi-Haradova choroba (kritéria 1 až 3, a to buď 4 nebo 5 musí být přítomen)
- Žádná historie pronikající oční trauma nebo chirurgický zákrok předchází počáteční nástup uveitida, a
- Žádné klinické nebo laboratorní známky svědčící pro jiné oční onemocnění subjekty, a
- Bilaterální oční zapojení.
- Neurologické/sluchové nálezy; jak je definován pro kompletní Vogtův-Koyanagiho-Haradova choroba výše, nebo
- Kožní nálezy, jak je definován pro kompletní Vogtův-Koyanagiho-Haradova choroba výše.
Pravděpodobné, Vogt-Koyanagi-Haradova choroba (izolované oční onemocnění; kritéria 1 až 3 musí být přítomen)
- Žádná historie pronikající oční trauma nebo chirurgický zákrok předchází počáteční nástup uveitidy.
- žádné klinické nebo laboratorní důkazy svědčící o jiných entitách očního onemocnění.
- Bilaterální oční zapojení, jak je definován pro kompletní Vogtův-Koyanagiho-Haradova choroba výše. „
Testování/Laboratorní Práce
V počáteční vyšetření z VKH, jeden by měl zvážit získání následujících testů:
- Optická koherenční tomografie (OCT): V akutní uveitické fázi bude OCT pravděpodobně vykazovat významné choroidální zahušťování a serózní oddělení sítnice. V ny tekutiny nahromadění může mít septations věřil být fibrinové membrány a zánětlivých produktů, vytvoření zavěšené konstrukce, který může být také vidět na fluorescenční angiografie. V rekonvalescentní fázi může OCT detekovat oblasti ztenčení sítnice po odeznění zánětu po léčbě kortikosteroidy .
- B-scan ultrasonografie: V akutní fázi může ultrasonografie vykazovat difúzní zadní choroidální zahušťování, zadní sklerální zahušťování, oddělení sítnice a sklovité opacity . Při ultrazvukové biomikroskopii lze pozorovat ciliární výpotky . Tento test je také užitečný pro vyloučení zadní skleritidy.
- Fluorescenční angiografie (FA): Klasicky, FA odhaluje multifokální choroidální hypofluorescent tečky v rané fázi, po níž následuje více kontaktních hyperfluorescent oblasti s difúzní úniku v pozdní fázi . Barvivo uniká RPE a hromadí se v subretinálním prostoru obklopujícím hyperfluorescentní tečky. FA může být diagnosticky užitečné, když VKH onemocnění představuje bez extraokulárních příznaků. Ve střední periferii lze pozorovat hyperfluorescenci optického disku a defekty okna způsobené atrofickými chorioretinálními jizvami . FA v chronické recidivující fázi onemocnění VKH vykazuje nespecifické defekty okna v důsledku poškození RPE, choroidální neovaskularizace a subretinální fibrózy .
- Indokyaninová zelená (ICG) angiografie: Rané fázi ICG líčí hyperfluorescent stromální cév, které ukazují, choroidální vaskulopatie a hypofluorescent tmavé tečky, které odpovídají granulomy a zpoždění nerovnoměrné plnění choroidálních cév . Pozdní fáze odhaluje fuzzy stromální vaskulární vzory a difúzní choroidální hyperfluorescenci. Hyperfluorescence disku naznačuje závažné onemocnění. ICGA může detekovat subklinický choroidální zánět ve velmi raných stádiích nebo dokonce po systémové terapii .
- lumbální punkce: pleocytóza v mozkomíšním moku je přítomna u většiny pacientů s VKH. Lumbální punkce by mělo být provedeno brzy v průběhu nemoci od pleocytózou může vyřešit
Léčba/Management/Pokyny
Léčba cíle v VKH patří včasná diagnóza a aktivní potlačení zánětu, spolu s prevenci recidivující zánět a vize ohrožující komplikací, jako je glaukom, bulózní odchlípení sítnice, choroidální neovaskularizace.
systémová léčba kortikosteroidy je preferovanou terapií onemocnění VKH, zejména během akutního uveitického stadia. Bylo prokázáno, že trasa kortikosteroidů podání (perorální versus intravenózní) nemá vliv na zrakovou ostrost nebo výskyt vizuálně-významné komplikace v léčbě akutní VKH . U závažného onemocnění je doporučeným protokolem intravenózní podávání methylprednisolonu po dobu tří dnů s následnou perorální léčbou vysokou dávkou prednisonu. U mírného až středně závažného onemocnění může být vysoká dávka perorálního prednisonu dostatečná při dávce 1-2 mg / kg / den. Dávka steroidů by měla být pomalu snižována přibližně po dobu šesti měsíců, aby se zabránilo opakování . Agresivní včasná léčba, spolu se sériovým testováním FA, které ukazuje zmizení úniku barviva RPE, může pomoci zabránit další progresi onemocnění, recidivě a extraokulárním projevům . Lokální steroidy a cykloplegika mohou snížit buňky v přední komoře a sklivce.
do sklivce a sub-Čep injekce triamcinolonu byly použity pro krátkodobou kontrolu nitroočního zánětu při akutní nebo opakující se fáze; tyto lokální terapie by měla být zvážena v případě vzpurné onemocnění a u pacientů, kteří špatně snášejí nepříznivé systémové vedlejší účinky steroidů vzhledem k rozšířené steroid kužel. Intravitreální anti-VEGF injekce se někdy používají ke kontrole choroidální neovaskularizace a v případech perzistentních foveálních serózních oddělení sítnice .
Steroid-šetřící činidla, např. antimetabolity, inhibitory kalcineurinu, biologických materiálů, TNF-alfa inhibitory, nebo cytotoxické látky mohou být použity k léčbě VKH a měla by být pečlivě sledována, často ve spolupráci s revmatologie služby . Probíhala diskuse o použití nesteroidních imunosupresiv jako první linie léčby onemocnění VKH. Nedávná studie však neodhalila žádné rozdíly ve výsledcích mezi časnou imunomodulační léčbou první linie (IMT) a samotnou léčbou prednisonem . Dále jsou imunosupresivní a biologické terapie drahé a vyžadují pečlivé hodnocení před léčbou a časté sledování krevního obrazu k posouzení závažných vedlejších účinků.
v chronicko-recidivujícím stádiu může častá recidiva naznačovat rezistenci na léčbu kortikosteroidy a naznačuje potřebu imunomodulační léčby šetřící steroidy . Výhodným činidlem pro recidivu rezistentní na steroidy nebo intoleranci steroidů je cyklosporin . Infliximab, rituximab, adalimumab a interferon alfa-2a jsou biologická činidla, která byla také použita k léčbě refrakterní uveitidy u onemocnění VKH.
K léčbě přední uveitidy často spojené s akutní VKH, lokální steroidy (např. prednisolon acetát 1%) a lokální cyloplegia (např. cyklopentolát 1% nebo atropin 1%), by měly být stanoveny v závislosti na stupni zánět přední oční komory.
oční komplikace jsou běžně spojeny s onemocněním VKH. Vzhledem k více stádiím a různým prezentacím, ve kterých může pacient s VKH prezentovat, může být léčba v mnoha případech zpožděna. U těžkých forem VKH a při recidivách může být nitrooční zánět obtížně kontrolovatelný a může vést ke strukturálnímu poškození. Více než 50% pacientů vyvinout komplikace, včetně katarakty, sekundárního glaukomu, choroidální neovaskulární membrány, ny fibróza, nebo kombinace těchto (Obrázek 6) .
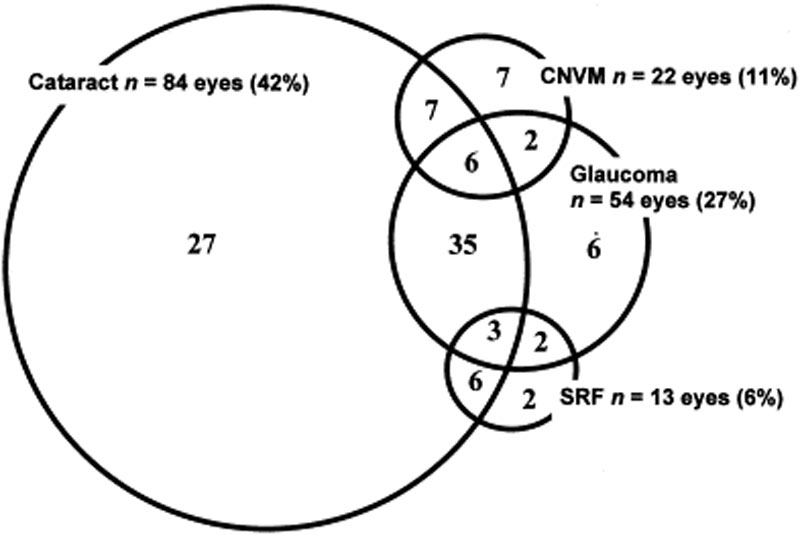
obrázek 6: Vennův diagram ukazující komplikace u pacientů s VKH. (Používá se se svolením Am J Oftalmol. 2001;131(5):599-606 )
EPIDEMIOLOGIE A ETIOLOGIE
|
SIGNS
|
PŘÍZNAKY
|
LÉČBA/MANAGEMENT
|
- Du L, Kijlstra A, Yang P. Vogt-Koyanagi-Harada disease: nové poznatky o patofyziologii, diagnostice a léčbě. Prog Retin Eye Res 2016; 52: 84-111. https://PubMed.gov/26875727. Doi: 10.1016 / j. preteyeres.2016.02.002
- Yamaki K, Gocho K, Hayakawa K, Kondo jsem, Sakuragi. S. Tyrosinase rodiny proteinů jsou antigeny specifické pro Vogt-Koyanagi-Haradova choroba. J Immunol 2000; 165 (12): 7323-7329. https://PubMed.gov/11120868
- Horie Y, Takemoto Y, Miyazaki, Namba K, Kase S, Yoshida K, Ota M, Hasumi Y, Inoko H, Mizuki N, Ohno S. Rodina genů tyrosinázy a Vogt-Koyanagi-Harada nemoc u japonských pacientů. Mol Vis 2006; 12: 1601-1605. https://PubMed.gov/17200659
- ng JY, Luk FO, Lai TY, Pang CP. Vliv molekulární genetiky na Vogtovu-Koyanagiho-Haradovu chorobu. Jaromír Jágr 2014; 4: 20. https://PubMed.gov/25097674. DOI: 10.1186 / s12348-014-0020-1
- Bowling B. Uveitis. Kanski ‚ s Clinical Ophthalmology New York, New York: Elsevier; 2016; Kapitola 11; s. 395-465.
- Yeh PT YC, Yang CH, Lin CP. Nonrhegmatogenní Odchlípení Sítnice. V: Schachat AP SS, Hinton DR, Wilkinson CP, Wiedemann P,, editor. Ryanova sítnice. New York: Elsevier; 2018; kapitola 99; p. 1828-1849.
- Goto H RK, Rao N. Vogt–Koyanagi-Harada nemoc. In: Schachat AP SS, Hinton DR, Wilkinson CP, Wiedemann P, editor. Ryanova sítnice. New York, New York: Elsevier; 2018; kapitola 78; s. 1505-1515.
- Riddington L, Hall aj, Tait B, Nicholson I, Varney m. Vogt-Koyanagi-Harada syndrom u pacientů vietnamského původu. Aust N Z J Ophthalmol 1996; 24 (2): 147-149. https://PubMed.gov/9199747
- Sugita S, Takase H, Kawaguchi T, Taguchi C, Mochizuki m. Křížové reakce mezi tyrosinase peptidy a cytomegalovirus antigenu T buněk od pacientů s Vogt-Koyanagi-Haradova choroba. Int Oftalmol 2007; 27 (2-3): 87-95. https://PubMed.gov/17253112. DOI: 10.1007 / s10792-006-9020-y
- Freund BK SD, Mieler WF, Yannuzzi LA. Zánět. Atlas Sítnice. New York, New York: Elsevier 2017; Kapitola 4; p. 279-398.
- Rao N. Vogt-Koyanagi-Harada nemoc. V: J YMaD, editor. Oftalmologie. New York, New York: Elsevier; 2014; kapitola 7.17; s. 761-763.
- Rao NA, Xu S, Font RL. Sympatická oftalmie. Imunohistochemická studie epitelioidních a obřích buněk. Oftalmologie 1985; 92 (12): 1660-1662. https://PubMed.gov/4088616
- Nussenblatt RB. Vogtův-Koyanagiho – Haradův Syndrom. V: Whitcup RBNaSM, editor. Uveitida: základy a klinická praxe. 4. vydání ed: Elsevier; 2010; Kapitola Kapitola 24.
- Čtení RW, Holandsku GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L, Pivetti-Pezzi P, Tesla HH, Usui M. Revidovaná diagnostická kritéria pro Vogt-Koyanagi-Harada onemocnění: zpráva z mezinárodní výbor pro nomenklaturu. Am J Oftalmol 2001; 131 (5): 647-652. https://PubMed.gov/11336942
- Chung H, Choi DG. Klinická analýza uveitidy. Korean J Ophthalmol 1989; 3 (1): 33-37. https://PubMed.gov/2795939. Doi: 10.3341 / kjo.1989.3.1.33
- Abu El-Asrar AM, Al-Kharashi JAKO, Aldibhi H, Al-Fraykh H, Kangave D. Vogt-Koyanagi-Harada onemocnění u dětí. Oko (Lond) 2008;22(9):1124-1131. https://PubMed.gov/17479116. Doi: 10.1038 / sj.oko.6702859
- Martin TD, Rathinam SR, Cunningham ET. Prevalence, klinické charakteristiky a příčiny ztráty zraku u dětí s nemocí Vogt-Koyanagi-Harada v jižní Indii. Sítnice 2010; 30 (7): 1113-1121. https://PubMed.gov/20168275. DOI: 10.1097 / IAE.0b013e3181c96a87
- Forster DJ, Green RL, Rao NA. Jednostranný projev syndromu Vogt-Koyanagi-Harada u 7letého dítěte. Am J Oftalmol 1991; 111 (3): 380-382. https://PubMed.gov/2000916
- Yamamoto Y, Fukushima A, Nishino k, Koura Y, Komatsu T, Ueno h. Vogt-koyanagi-harada nemoc s nástupem u starších pacientů ve věku 68 až 89 let. Jpn J Oftalmol 2007; 51 (1): 60-63. https://PubMed.gov/17295144. DOI: 10.1007 / s10384-006-0379-0
- Wang Y, Chan CC. Genderové rozdíly u vogt-koyanagi-haradovy choroby a sympatické oftalmie. J Oftalmol 2014; 2014: 157803. https://PubMed.gov/24734166. DOI: 10.1155/2014/157803
- Nakao K, Abematsu N, Mizushima Y, T. Sakamoto Optického disku otok v Vogt-Koyanagi-Haradova choroba. Invest Ophthalmol Vis Sci 2012; 53 (4): 1917-1922. https://PubMed.gov/22408010. DOI: 10.1167/iovs.11-8984
- Rao NA, Gupta, Dustin L, Chee SP, Okada AA, Khairallah M, Bodaghi B, Lehoang P, Accorinti M, Mochizuki M, Prabriputaloong T, Číst RW. Frekvence rozlišování klinických příznaků u Vogt-Koyanagi-haradovy choroby. Ophthalmology 2010;117(3):591-599, 599.e591. https://PubMed.gov/20036008. DOI: 10.1016/j.ophtha.2009.08.030
- Veerappan M, Fleischman D, Ulrich JN, Stinnett SS, Jaffe GJ, Allingham RR. The Relationship of Vogt-Koyanagi-Harada Syndrome to Ocular Hypertension and Glaucoma. Ocul Immunol Inflamm 2017;25(6):748-752. https://PubMed.gov/27438521. DOI: 10.1080/09273948.2016.1189578
- Baltmr A, Lightman S, Tomkins-Netzer O. Vogt-Koyanagi-Harada syndrome – current perspectives. Clin Ophthalmol 2016;10:2345-2361. https://PubMed.gov/27932857. DOI: 10.2147/OPTH.S94866
- Kitaichi N, Matoba H, Ohno S. pozitivní roli lumbální punkcí v diagnostice Vogt-Koyanagi-Haradova choroba: podskupin lymfocytů ve vodné humor a mozkomíšního moku. Int Oftalmol 2007; 27 (2-3): 97-103. https://PubMed.gov/17211585. DOI: 10.1007/s10792-006-9016-7
- Oshima Y, Harino S, Hara Y, Tano Y. Indocyanine green angiografické nálezy v Vogt-Koyanagi-Haradova choroba. Am J Ophthalmol 1996; 122 (1): 58-66. https://PubMed.gov/8659599
- Čtení RW, Yu, F, Accorinti M, Bodaghi B, Chee SP, Fardeau C, Goto H, Holandsku GN, Kawashima H, Kojima E, Lehoang P, Lemaitre C, Okada AA, Pivetti-Pezzi P, Secchi, Viz RF, Tabbara KF, Usui M, Rao NA. Vyhodnocení účinku na výsledky způsobu podávání kortikosteroidů při akutní Vogt-Koyanagi-haradově chorobě. Am J Oftalmol 2006; 142 (1): 119-124. https://PubMed.gov/16815259. Doi: 10.1016 / j. ajo.2006.02.049
- Rubsamen PE, Gass JD. Vogtův-Koyanagiho – haradův syndrom. Klinický průběh, terapie a dlouhodobý vizuální výsledek. Arch Oftalmol 1991;109(5):682-687. https://PubMed.gov/2025171
- Urzua CA, Velasquez V, Sabat P, Berger O, Ramirez S, Goecke, Vásquez DH, Gatica H, Guerrero J. Dříve imunomodulační léčby je spojeno s lepší vizuální výsledky v podskupině pacientů s Vogt-Koyanagi-Haradova choroba. Acta Ophthalmol 2015; 93 (6): e475-480. https://PubMed.gov/25565265. Doi: 10.1111/aos.12648
- Read RW, Rechodouni A, Butani N, Johnston R, LaBree LD, Smith RE, Rao NA. Komplikace a prognostické faktory u Vogt-Koyanagiho-Haradovy choroby. Am J Oftalmol 2001; 131 (5): 599-606. https://PubMed.gov/11336934
Doporučené Citační Formát
Mai AP, Tran C, Wilson CW, Fox AR, Boldt VM. Vogt-Koyanagi-Harada (VKH) nemoc. EyeRounds.org. 1. Dubna 2019. Dostupné z http://EyeRounds.org/cases/284-vogt-koyanagi-harada.htm
Leave a Reply